環境: macOS Sierra 10.12.4, Gromacs 2016.3.
MacKerell Labで配布されているGROMACS用のCharmm力場ファイルのNovember 2016版に亜鉛に配位したシステイン用の残基(CYN)が追加されていたことに気付いたので(ただしパラメータは従来から存在するCYM残基と変更はない),これを使って亜鉛イオンを2原子配位したC1ドメインのトポロジーを作成してみました.
AMBER用のZAFF(Zinc AMBER Force Field)は配位パターンそれぞれに異なるパラメータを用意して,亜鉛イオンの正電荷とシステイン(thiolate form) の負電荷を錯体全体に分散させています.それに対して,Charmmに新たに残基として追加されているのは亜鉛に配位したシステイン(CYN) のみで, 亜鉛を含む結合,結合角,二面角のパラメータはありますが,ZN,CYN,HIS残基はそれぞれ+2,−1,0の電荷を持つ残基として表現されます.アプローチとしては,ZAFFの方が良さそうです.
硫黄–亜鉛間の距離はZAFF(CCCH型でHはHIE)が0.2355,Charmmが0.232,窒素–亜鉛間はZAFFが0.2176,Charmmが0.207でした.
タンパク質用トポロジー
MacKerell Labからcharmm36-nov2016.ff.tgzをダウンロードします.
$ tar xvzf charmm36-nov2016.ff.tgz
$ mv charmm36-nov2016.ff $GMXPREFIX/share/gromacs/top/
1PTR.pdb (PKCδ C1Bドメイン) をProtein Data Bankからダウンロードする.
欠けた側鎖を補完し,N末とC末にGly残基を追加する (亜鉛に配位するHISとCYSが末端に位置しないように).PymolとModellerを使用した方法については別記事を参照のこと.modeller.pdbとしてコピー.
1PTRファイル中のPRB残基 (phorbol 13-acetate) の行を分離して別ファイルにする (PRB.pdb).
Gromacsのpdb2gmxでPDBファイルを処理する際に,原子間で追加の結合を形成させる条件が書かれたspecbond.datを作業ディレクトリにコピーして編集します.
$ cp $GMXPREFIX/share/gromacs/top/specbond.dat .
specbond.datを以下のように編集します(2行追加して,1行目の数字を変更).7列目の数値(単位はnm)の±10%以内に2つの原子がある時に結合が形成されます.
10 CYS SG 1 ZN ZN 1 0.24 CYN ZN2 HIS ND1 1 ZN ZN 1 0.20 HSE ZN2 CYS SG 1 CYS SG 1 0.2 CYS2 CYS2 CYS SG 1 HEM FE 2 0.25 CYS2 HEME CYS SG 1 HEM CAB 1 0.18 CYS2 HEME CYS SG 1 HEM CAC 1 0.18 CYS2 HEME HIS NE2 1 HEM FE 1 0.2 HIS1 HEME MET SD 1 HEM FE 1 0.24 MET HEME CO C 1 HEME FE 1 0.19 CO HEME CYM SG 1 CYM SG 1 0.2 CYS2 CYS2
$ gmx pdb2gmx -f modeller.pdb -o conf.out -ignh力場は 9: CHARMM36 all-atom force field (November 2016),
水は1: TIP3P TIP 3-point, recommended, by default uses CHARMM TIP3 with LJ on H
を選びます.
Linking HIS-2 ND1-12 and ZN-53 ZN-410... Linking CYS-15 SG-129 and ZN-54 ZN-411... Linking CYS-18 SG-153 and ZN-54 ZN-411... Linking CYS-32 SG-257 and ZN-53 ZN-410... Linking CYS-35 SG-280 and ZN-53 ZN-410... Linking HIS-40 ND1-315 and ZN-54 ZN-411... Linking CYS-43 SG-342 and ZN-54 ZN-411... Linking CYS-51 SG-405 and ZN-53 ZN-410...
と出力され,結合が作られていることが確認できました.いつも通り,シミュレーションBOXを作成し,水を撒いて,イオンを加えて電荷を中和します.
$ gmx editconf -f conf.gro -bt cubic -d 1 -o box.gro
$ gmx solvate -cp box.gro -p topol.top -o solv.gro
$ gmx grompp -f minim.mdp -c solv.gro -o ion.tpr
$ gmx genion -s ion.tpr -p topol.top -o ion.gro -nname CL -neutralイオンと置換する分子はGroup 15 SOLを選択.
トポロジーファイル (topol.top) の最後を確認すると,SOLとCLが追加されていました.
[ molecules ] ; Compound #mols Protein 1 SOL 5840 CL 1
エネルギー最小化を実行してみます.
$ gmx grompp -f minim.mdp -c ion.gro -o em.tpr
$ gmx mdrun -v -deffnm em問題なく動きました.以後,MDシミュレーションを行う時は,residuetypes.datファイルをコピーしてきて,亜鉛をタンパク質として扱った方が何かと便利です.
錯体の電荷を分散させていないこの形式と,電荷を分散させた形式から得られた結果でどんな違いがあるかは検証が必要です.
リガンドを加える
元ファイルから分離していたPRB.pdbをAvogadroで開きます(β版の1.90.0ではなくver 1.2).Bond orderを修正して,水素結合を付加,MMFF94s力場で構造最適化した後,mol2形式で保存します.PRB.mol2ファイルをテキストエディタで開いて,2行目の*****をPRBへ,残基名のLIGをPRBへ書き換えます.
このmol2ファイルをCGenFF Serverへアップロードし,Charmm形式のトポロジーファイル (PRB.str) を得ます.上述したMacKerell Labで配布されているcgenff_charmm2gmx.pyを使って,GROMACS用トポロジーに変換します.
$ cgenff_charmm2gmx.py PRB PRB.mol2 PRB.str $GMXPREFIX/share/gromacs/top/charmm36-nov2016.ffこのパラメータを使って最適化した構造でも,DFT計算(M06-2X/6-311++G(d,p) レベル)で得られた構造と比較的一致していました(下図左: DFT, 右: CGenFF).
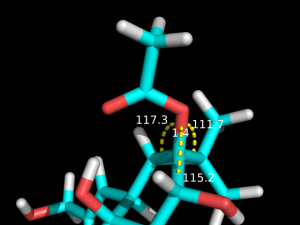
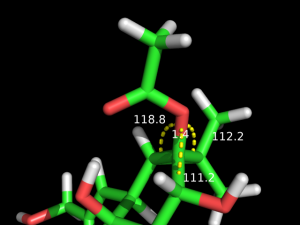
PRB.strファイルを見てみると,シクロプロパン環や橋頭位の炭素原子(CG3RC1)に結合した酸素原子(OG302)が関与する結合角のパラメータが炭素-炭素-炭素のパラメータから流用されていたので,これらの結合角のパラメータを修正しました.
Atom typesは
- CG3C31 – cyclopropyl carbon
- CG3RC1 – bridgehead in bicyclic systems containing at least one 5-membered or smaller ring
- OG302 – ester -O-
です.
[ bondtypes ] [ angletypes ] ; i j k func theta0 ktheta ub0 kub CG311 CG3RC1 OG302 5 113.500000 488.272800 0.25610000 9338.69 CG321 CG3RC1 OG311 5 113.500000 488.272800 0.25610000 9338.69 CG3C31 CG3RC1 OG302 5 111.000000 446.432800 0.25610000 6694.40 CG3RC1 CG3RC1 OG302 5 111.000000 446.432800 0.25610000 6694.40 以下のように修正 CG311 CG3RC1 OG302 5 110.000000 633.457600 0.00000000 0.00 CG321 CG3RC1 OG311 5 113.500000 633.457600 0.00000000 0.00 CG3C31 CG3RC1 OG302 5 111.000000 633.457600 0.00000000 0.00 CG3RC1 CG3RC1 OG302 5 114.500000 633.457600 0.00000000 0.00
Open Babelを使って生成されたprb_ini.pdbをgromacs形式に変換します.
$ obabel -ipdb prb_ini.pdb -Oprb_ini.gro
前節の処理途中のconf.groの冒頭の原子数にPRB分子の原子数を足して(805→864),ファイルの最後にprb_ini.gro中の座標部分をくっつけます (conf_ligand.groとして保存).
途中から
53ZN ZN 804 2.174 1.867 4.287
54ZN ZN 805 1.196 1.317 3.294
1PRB C1 1 1.345 2.964 2.324
1PRB C2 2 1.438 3.077 2.292
1PRB O1 3 1.227 3.011 2.371
(中略)
1PRB H29 58 1.137 2.370 2.862
1PRB H30 59 1.319 2.314 2.651
2.74916 2.64127 2.90824
topol.topの最初の方でリガンドのトポロジーを読み込むか,書き込みます.最後の [ molecules ] セクションにPRB分子を加えます.
; Include forcefield parameters #include "charmm36-nov2016.ff/forcefield.itp" #include "prb.prm" #include "prb.itp" [ moleculetype ] ; Name nrexcl Protein 3 (中略) [ molecules ] ; Compound #mols Protein 1 PRB 1
$ gmx editconf -f conf_ligand.gro -bt cubic -d 1 -o box_ligand.gro
$ gmx solvate -cp box_ligand.gro -p topol.top -o solv_ligand.gro
$ gmx grompp -f minim.mdp -c solv_ligand.gro -o ion.tpr以下のようなエラーが出ます.
ERROR 1 [file prb.prm, line 81]: Encountered a second block of parameters for dihedral type 9 for the same atoms, with either different parameters and/or the first block has multiple lines. This is not supported.
CGenFF serverの力場のバージョンは3.0.3ですが,charmm36-nov2016.ffに含まれるCGenFFのバージョンは4.0にアップデートされているので,パラメータが重複していました.prb.prmファイルの81-83行目をコメントアウトか削除します.
$ gmx grompp -f minim.mdp -c solv.gro -o ion.tpr
$ gmx genion -s ion.tpr -p topol.top -o ion_ligand.gro -nname CL -neutral
$ gmx grompp -f minim.mdp -c ion_ligand.gro -o em_ligand.tpr
$ gmx mdrun -v -deffnm em_ligand
$ gmx trjconv -f em_ligand.gro -s em_ligand.tpr -o em_ligand.pdb
(了)