研究室内メモ
環境: CentOS 7, GROMACS 2019.5.
下の文献のシミュレーションの最初の部分だけやってみました。
Ryckbosch, S. M.; Wender, P. A.; Pande, V. S. Molecular dynamics simulations reveal ligand-controlled positioning of a peripheral protein complex in membranes. Nat. Commun. 2017, 8, 6. DOI: 10.1038/s41467-016-0015-8.
論文中の分子動力学(MD)シミュレーションの手順は
- PKCδ C1Bドメインとリガンド(Bryostatin-1, Wender’s bryolog, PDBu, prostratin)の複合体あるいはドメインのみを溶液中から脂質二重膜(POPS 128分子)へZ軸に沿って200 nsかけて引っ張り上げる。引っ張るのはポケット形成するアミノ酸残基(全体の約3分の1)と(存在するなら)リガンド。
- 軌跡(トラジェクトリ)から反応座標に沿って100個の初期座標を取る。
- それぞれの初期座標を使ったシミュレーションを40クローン(20 ns)行う。
- これらのシミュレーションから、タンパク質リガンド複合体の膜に対する距離と角度で二次元プロファイルを得る。この配置を全て網羅するように新たに1000個の初期座標を取る。
- この1000個の初期座標 × 4クローン × 30 ns秒のシミュレーションを行う。
- マルコフ状態モデル(MSM)による自由エネルギー解析
全て合わせたシミュレーション時間はBryostatin-1、PDBu、タンパク質のみが400マイクロ秒、prostratinが580マイクロ秒、Bryologが300マイクロ秒。途方もなく長い。
計算プログラム、力場: プログラム, GROMACS 4.5.3; 計算機, Folding@home; 力場 –
AMBER99SB-ILDN(タンパク質), GAFF2(リガンド), Slipids(脂質), TIP3P(水)。
普通、C1ドメイン-リガンド複合体を脂質二重膜に埋め込んだ初期構造をPPM serverとCHARMM-GUIで作るとタンパク質が埋まった側(inner leaflet)のリン脂質分子数が6個程少くなります。上下の分子数の差を引っ張り上げる時にどうしているのかと思ったら、outer leafletとinner leafletのリン脂質分子数は同じ(64分子ずつ)で無理矢理タンパク質リガンド複合体を引っ張り上げていました。そこの細かいところは気にしていないようです。
引っ張り上げるところだけならまず出来そうだと思ってPKCδ C1BドメインとTPAの複合体でやってみることにしました。TPAのような疎水性が高い分子ははじめから膜中に分配していると思いますが、細かいことは気にせずやってみます。
COM-pulling
ファイル
COMは “center of mass” 「質量中心」の略称です。CHARMM-GUIサーバを使って1PTR.pdbから作成したPKCδ-C1BドメインとTPAの複合体構造をPOPS二重膜の少し下側に配置しました。CHARMM-GUIが出力する通常の平衡化(step6.6まで)を行った後、step6.6の条件(タンパク質とリガンドの重原子に位置拘束がかかっている)でさらに100 ns平衡化を行いました。次に、mdpファイルに以下のようにpullingのための記述を加えてCOM-pullingシミュレーションを実行しました。
; Pull code pull = yes pull_ncoords = 1 ; 反応座標は1つだけ pull_ngroups = 2 ; 2つのグループで反応座標を定義 pull_group1_name = TPA ; リガンド pull_group2_name = MEMB ; 脂質二重膜全体 pull_coord1_type = umbrella ; harmonic potential pull_coord1_geometry = distance ; 距離を変化させる pull_coord1_dim = N N Y ; Z軸に沿ってpullする pull_coord1_groups = 1 2 pull_coord1_start = yes ; define initial COM distance > 0 pull_coord1_rate = -0.00025 ; -0.00025 nm per ps = -0.25 nm(2.5 Å)per ns. pull_coord1_k = 1000 ; kJ mol^-1 nm^-2
結果
水中では丸まっていたTPAの側鎖が膜中では伸びたのが印象的でした。トラジェクトリから初期座標を取って、反応座標を固定してそれぞれMDシミュレーションをした後(アンブレラサンプリング)、WHAM(weighted histogram analysis method)法で解析することで反応座標上のPMF(potential of mean force)曲線(反応座標に沿って自由エネルギーをプロットしたもの)を得ることができるそうです。
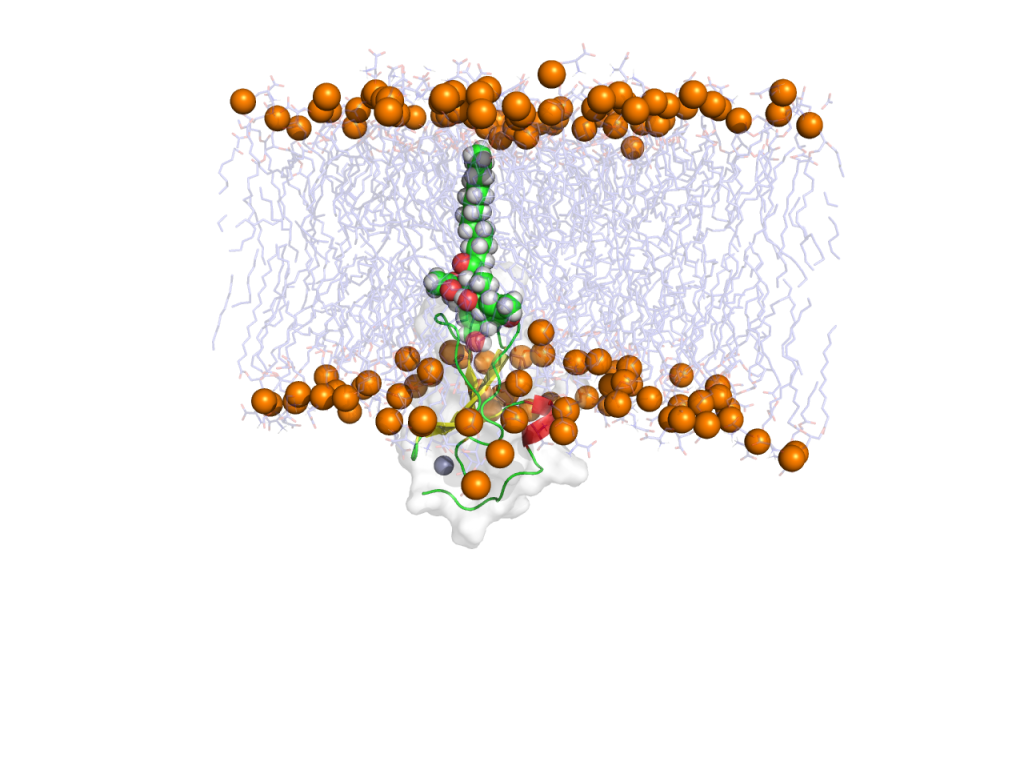
画像
橙色の球はリン脂質のリン原子を表わす。
最初
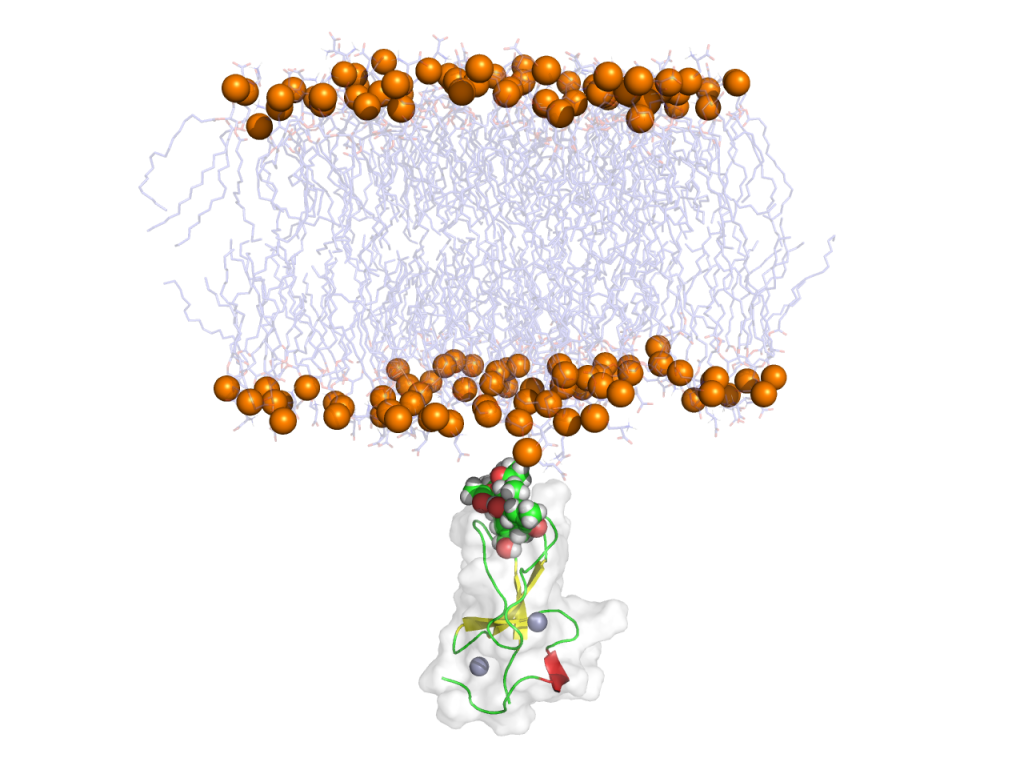
最後
動画
(了)
一般的な質問で恐縮ですが、topologyファイル(pdb, gro)で構造上の不備(ボンドが切れている、オーバーラップが有る、等)をチェックする方法は有りますでしょうか。膜構造の場合、これに起因するエラーが多いのでお尋ねしました。
機械的にチェックする方法は残念ながら知りません。申し訳ありません。
膜の脂質鎖部分がオーバーラップして、エネルギー最小化が止まるエラーに遭遇することはあります。そういった場合は、該当原子のGROファイル中の座標のxかyかzの値を少し書き換えて位置をずらしてから、エネルギー最小化を行っています。