PKC C1ドメインとphorbol-13-acetate複合体のMDシミュレーション
GromacsでPKC C1ドメインとリガンドの複合体のMDシミュレーションを行うためのメモです.
力場
- タンパク質: AMBER ff14SB + ZAFF
- リガンド: GAFF2
- 水: TIP3P
参考サイト: Ag++: antechamberを使ってGaussian 09からAMBER用の力場・電荷パラメータを作成する
トポロジーファイルの作成
タンパク質ファイルの側鎖補完,残基付加,修正については,別ページと同じ.modeller.pdbからリガンド座標をPRB.pdbとして一旦分離.Avogadroでリガンド座標ファイル (PRB.pdb) を開き,結合の不飽和度を修正した後,水素を付加,MMFF94s力場で最適化,pdb形式で保存する.重原子と水素原子で残基名と残基番号が異なる場合は統一する(重要.Avogadroで付加した水素原子の見出しはHETATM,残基名はLIG,残基番号は1になっている).
$ antechamber -fi pdb -fo prepi -i PRB.pdb -o PRB.prep -c bcc -at gaff2
$ parmchk2 -i PRB.prep -o PRB.frcmod -f prepi -s gaff2
$ tleap
> source leaprc.gaff2
> loadamberprep PRB.prep
> loadamberparams PRB.frcmod
> mol = loadpdb NEWPDB.PDB
> saveamberparm mol PRB.prmtop PRB.inpcrd
> quitAntechamberの実行には1分半程かかる.Amber16付属のParmEdを使って,AMBER形式のファイルをGROMACS形式へ変換する.
$ python
>>> import parmed as pmd
>>> amber = pmd.load_file('PRB.prmtop',xyz='PRB.inpcrd')
>>> amber.save('gromacs.top')
>>> amber.save('gromacs.gro')
>>> quit()ここで作成したトポロジーが正常かどうか,Gromacsでエネルギー最小化計算を実行してみる.
$ gmx grompp -f em.mdp -c gromacs.gro -p gromacs.top -o em.tpr
$ gmx mdrun -v -deffnm emem.groをpymolで開いてみると,13位周辺が歪んだ構造になってしまっている.PRB_GMX.topをテキストエディタで開いて,13位酸素原子 (今回はOA1) に関するパラメータを見てみると,
[ bonds ]
52 53 1 0.14370 233885.600000
[ angles ]
48 52 53 1 115.68005 693.590048
27 52 53 1 59.09003 975.641856
28 52 53 1 59.09003 975.641856
2箇所のangleが59.09°となっていて,どうやらシクロプロパン環の炭素原子 (atom type: cx) に結合した13位酸素原子 (atom type: os) はepoxideの酸素原子と判定しているらしい (エステルとエーテルのsp3酸素原子のatom typeは同じ).また,
1-4: angle 28 52 duplicates bond ('triangular' bond) or angle ('square' bond)
1-4: angle 27 52 duplicates bond ('triangular' bond) or angle ('square' bond)
1-4: angle 27 28 duplicates bond ('triangular' bond) or angle ('square' bond)
というWarningが出る.シクロプロパン環があると出るWarningとのこと (2つのアングルを指定すれば,後1つは必要ないため).
シクロプロパン環に結合したエステル基のパラメータがないので,この部分の角度については計算値を入れることにした.
BabelでGaussianのinputファイルに変換した後,headerを修正して構造最適化を実行.
$ babel PRB.pdb PRB.com# opt wb97xd/6-31g(d) geom=connectivity
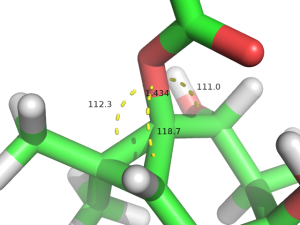 DFT計算での最適化構造の13位周りの角度と距離は右図のようになった.この値をトポロジーファイルに入れた.
DFT計算での最適化構造の13位周りの角度と距離は右図のようになった.この値をトポロジーファイルに入れた.
修正後:
48 52 53 1 111.0 693.590048
27 52 53 1 118.7 693.590048
28 52 53 1 112.3 693.590048
もう一度Gromacsで最適化計算を行ったところ,正常な構造になっていた.
水素が付加されたリガンドの座標が含まれるPDBファイルを1PTR_ZAFF_PRB.pdbとします.
ATOM 408 C GLY 52 28.447 17.465 44.064 1.00 47.64 C ATOM 409 O GLY 52 27.783 18.409 43.643 1.00 47.64 O TER HETATM 410 ZN ZN2 53 21.740 18.670 42.875 1.00 40.35 ZN TER HETATM 411 ZN ZN2 54 11.959 13.171 32.940 1.00 47.44 ZN TER ATOM 1 O3 PRB 1 7.931 22.364 24.610 -0.519100 ATOM 2 C3 PRB 1 8.750 23.087 24.056 0.544300 ATOM 3 C2 PRB 1 9.135 22.950 22.639 -0.222400 ATOM 4 C19 PRB 1 8.565 21.929 21.737 -0.039900 ATOM 5 H24 PRB 1 8.997 21.999 20.733 0.050367
AmberToolsに含まれるtleap用のinputファイルを作成 (1PTR_ZAFF_PRB_tleap.in)
source leaprc.protein.ff14SB #Source the ff14SB force field
source leaprc.water.tip3p #Source water parameters
source leaprc.gaff2 #Source the GAFF force field
addAtomTypes { { "ZN" "Zn" "sp3" } { "S2" "S" "sp3" } { "N1" "N" "sp3" } } #Add atom types for the ZAFF metal center with Center ID 2
loadamberprep ZAFF.prep #Load ZAFF prep file
loadamberparams ZAFF.frcmod #Load ZAFF frcmod file
loadamberprep ./PRB.prep
loadamberparams ./PRB.frcmod
mol = loadpdb 1PTR_ZAFF_PRB.pdb #Load the PDB file
bond mol.53.ZN mol.32.SG #Bond zinc ion with SG atom of residue CYM31
bond mol.53.ZN mol.35.SG #Bond zinc ion with SG atom of residue CYM34
bond mol.53.ZN mol.51.SG #Bond zinc ion with SG atom of residue CYM50
bond mol.53.ZN mol.2.ND1 #Bond zinc ion with SG atom of residue HIE1
bond mol.54.ZN mol.15.SG #Bond zinc ion with SG atom of residue CYM14
bond mol.54.ZN mol.18.SG #Bond zinc ion with SG atom of residue CYM17
bond mol.54.ZN mol.43.SG #Bond zinc ion with SG atom of residue CYM42
bond mol.54.ZN mol.40.ND1 #Bond zinc ion with SG atom of residue HIE39
savepdb mol 1PTR_ZAFF_PRB_dry.pdb #Save the pdb file
saveamberparm mol 1PTR_ZAFF_PRB_dry.prmtop 1PTR_ZAFF_PRB_dry.inpcrd #Save the topology and coordiante files
solvatebox mol TIP3PBOX 10.0 #Solvate the system using TIP3P water box
addions mol CL 0 #Neutralize the system using Cl- ions
savepdb mol 1PTR_ZAFF_PRB_solv.pdb #Save the pdb file
saveamberparm mol 1PTR_ZAFF_PRB_solv.prmtop 1PTR_ZAFF_PRB_solv.inpcrd #Save the topology and coordiante files
quit #Quit tleap
tleapを実行.
$ tleap -s -f 1PTR_ZAFF_PRB_tleap.in > 1PTR_ZAFF_PRB_tleap.outすぐ終了.
$ python
>>> import parmed as pmd
>>> amber = pmd.load_file('1PTR_ZAFF_PRB_solv.prmtop',xyz='1PTR_ZAFF_PRB_solv.inpcrd')
>>> amber.save('1PTR_ZAFF_PRB_solv.gro')
>>> amber.save('1PTR_ZAFF_PRB_solv.top')
>>> quit()GROMACS用座標ファイル (1PTR_ZAFF_PRB_solv_GMX.gro) とGROMACS用トポロジーファイル (1PTR_ZAFF_PRB_solv_GMX.top) が生成する.
上で説明したように,リガンド部分のtopologyを書き換える.
エネルギー最小化
minim.mdp (以下のinput filesはGROMACS Tutorial: Protein-Ligandより)
integrator = steep ; Algorithm (steep = steepest descent minimization) emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm emstep = 0.01 ; Energy step size nsteps = 50000 ; Maximum number of (minimization) steps to perform nstlist = 1 ; Frequency to update the neighbor list and long range forces ns_type = grid ; Method to determine neighbor list (simple, grid) rlist = 1.0 ; Cut-off for making neighbor list (short range forces) coulombtype = PME ; Treatment of long range electrostatic interactions rcoulomb = 1.0 ; Short-range electrostatic cut-off rvdw = 1.0 ; Short-range Van der Waals cut-off pbc = xyz ; Periodic Boundary Conditions (yes/no)
$ gmx grompp -minim.mdp -c 1PTR_ZAFF_PRB_solv_GMX.gro -p 1PTR_ZAFF_PRB_solv_GMX.top -o em.tpr
$ gmx mdrun -v -deffnm emIndexファイルの作成
$ gmx make_ndx -f em.gro
0 System : 11677 atoms
1 Protein : 709 atoms
2 Protein-H : 354 atoms
3 C-alpha : 44 atoms
4 Backbone : 132 atoms
5 MainChain : 177 atoms
6 MainChain+Cb : 215 atoms
7 MainChain+H : 222 atoms
8 SideChain : 487 atoms
9 SideChain-H : 177 atoms
10 Prot-Masses : 709 atoms
11 non-Protein : 10968 atoms
12 Other : 155 atoms
13 HE1 : 34 atoms
14 CY2 : 60 atoms
15 ZN2 : 2 atoms
16 PRB : 59 atoms
17 CL : 1 atoms
18 Ion : 1 atoms
19 HE1 : 34 atoms
20 CY2 : 60 atoms
21 ZN2 : 2 atoms
22 PRB : 59 atoms
23 CL : 1 atoms
24 Water : 10812 atoms
25 SOL : 10812 atoms
26 non-Water : 865 atoms
27 Water_and_ions : 10813 atoms
> 1 | 13 | 14 | 15 #タンパク質,亜鉛イオンをmerge
> 28 | 16 #タンパク質,亜鉛イオン,リガンドをmerge
> r 1-7 | r 15-21 | r 26-27 | r 29-54 #リガンドから遠い残基をグループ化
> qタンパク質-亜鉛-リガンド,リガンドから遠いタンパク質,リガンドそれぞれに対してrestraintファイルを作成
$ gmx genrestr -f em.gro -n index.ndx -o posre.itp -fc 1000 1000 1000
> 29
$ gmx genrestr -f em.gro -n index.ndx -o posre_far.itp -fc 1000 1000 1000
> 30トポロジーファイル (1PTR_ZAFF_PRB_solv_GMX.top) にposition restraintファイルの情報を書き込む.
829 820 823 824 4 180.00 4.60240 2 ; C7- C5- C6- C20 861 860 859 858 4 180.00 4.60240 2 ; CA2- OA2- CA1- OA1 #ifdef POSRES #include "posre.itp" #endif #ifdef POSRES_FAR #include "posre_far.itp" #endif [ moleculetype ] ; molname nrexcl ; TIP3P model WAT 2
NVT equilibration
nvt.mdp
define = -DPOSRES ; position restrain the protein and ligand ; Run parameters integrator = md ; leap-frog integrator nsteps = 50000 ; 2 * 50000 = 100 ps dt = 0.002 ; 2 fs ; Output control nstxout = 500 ; save coordinates every 1.0 ps nstvout = 500 ; save velocities every 1.0 ps nstenergy = 500 ; save energies every 1.0 ps nstlog = 500 ; update log file every 1.0 ps energygrps = Protein_HE1_CY2_ZN2 PRB ; Bond parameters continuation = no ; first dynamics run constraint_algorithm = lincs ; holonomic constraints constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained lincs_iter = 1 ; accuracy of LINCS lincs_order = 4 ; also related to accuracy ; Neighborsearching cutoff-scheme = Verlet ns_type = grid ; search neighboring grid cells nstlist = 10 ; 20 fs, largely irrelevant with Verlet rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm) rvdw = 1.4 ; short-range van der Waals cutoff (in nm) ; Electrostatics coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics pme_order = 4 ; cubic interpolation fourierspacing = 0.16 ; grid spacing for FFT ; Temperature coupling tcoupl = V-rescale ; modified Berendsen thermostat tc-grps = Protein_HE1_CY2_ZN2_PRB Water_and_ions ; two coupling groups - more accurate tau_t = 0.1 0.1 ; time constant, in ps ref_t = 300 300 ; reference temperature, one for each group, in K ; Pressure coupling pcoupl = no ; no pressure coupling in NVT ; Periodic boundary conditions pbc = xyz ; 3-D PBC ; Dispersion correction DispCorr = EnerPres ; account for cut-off vdW scheme ; Velocity generation gen_vel = yes ; assign velocities from Maxwell distribution gen_temp = 300 ; temperature for Maxwell distribution gen_seed = -1 ; generate a random seed
$ gmx grompp -f nvt.mdp -c em.gro -p 1PTR_ZAFF_PRB_solv_GMX.top -n index.ndx -o nvt.tpr
$ gmx mdrun -v -deffnm nvt
$ gmx energy -f nvt.edr -o temperature.xvg10分程で計算終了.
NPT equilibration
npt.mdp
define = -DPOSRES ; position restrain the protein and ligand ; Run parameters integrator = md ; leap-frog integrator nsteps = 50000 ; 2 * 50000 = 100 ps dt = 0.002 ; 2 fs ; Output control nstxout = 500 ; save coordinates every 1.0 ps nstvout = 500 ; save velocities every 1.0 ps nstenergy = 500 ; save energies every 1.0 ps nstlog = 500 ; update log file every 1.0 ps energygrps = Protein_HE1_CY2_ZN2 PRB ; Bond parameters continuation = yes ; first dynamics run constraint_algorithm = lincs ; holonomic constraints constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained lincs_iter = 1 ; accuracy of LINCS lincs_order = 4 ; also related to accuracy ; Neighborsearching cutoff-scheme = Verlet ns_type = grid ; search neighboring grid cells nstlist = 10 ; 20 fs, largely irrelevant with Verlet rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm) rvdw = 1.4 ; short-range van der Waals cutoff (in nm) ; Electrostatics coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics pme_order = 4 ; cubic interpolation fourierspacing = 0.16 ; grid spacing for FFT ; Temperature coupling tcoupl = V-rescale ; modified Berendsen thermostat tc-grps = Protein_HE1_CY2_ZN2_PRB Water_and_ions ; two coupling groups - more accurate tau_t = 0.1 0.1 ; time constant, in ps ref_t = 300 300 ; reference temperature, one for each group, in K ; Pressure coupling pcoupl = Parrinello-Rahman ; pressure coupling is on for NPT pcoupltype = isotropic ; uniform scaling of box vectors tau_p = 2.0 ; time constant, in ps ref_p = 1.0 ; reference pressure, in bar compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1 refcoord_scaling = com ; Periodic boundary conditions pbc = xyz ; 3-D PBC ; Dispersion correction DispCorr = EnerPres ; account for cut-off vdW scheme ; Velocity generation gen_vel = no ; velocity generation off after NVT
$ gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -p 1PTR_ZAFF_PRB_solv_GMX.top -n index.ndx -o npt.tpr
$ gmx mdrun -v -deffnm npt
$ gmx energy -f npt.edr -o pressure.xvgProduction MD
md.mdp
; Run parameters integrator = md ; leap-frog integrator nsteps = 500000 ; 2 * 500000 = 1000 ps (1 ns) dt = 0.002 ; 2 fs ; Output control nstxout = 0 ; suppress .trr output nstvout = 0 ; suppress .trr output nstenergy = 5000 ; save energies every 10.0 ps nstlog = 5000 ; update log file every 10.0 ps nstxout-compressed = 5000 ; write .xtc trajectory every 10.0 ps compressed-x-grps = System energygrps = Protein_HE1_CY2_ZN2 PRB ; Bond parameters continuation = yes ; first dynamics run constraint_algorithm = lincs ; holonomic constraints constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained lincs_iter = 1 ; accuracy of LINCS lincs_order = 4 ; also related to accuracy ; Neighborsearching cutoff-scheme = Verlet ns_type = grid ; search neighboring grid cells nstlist = 10 ; 20 fs, largely irrelevant with Verlet rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm) rvdw = 1.4 ; short-range van der Waals cutoff (in nm) ; Electrostatics coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics pme_order = 4 ; cubic interpolation fourierspacing = 0.16 ; grid spacing for FFT ; Temperature coupling tcoupl = V-rescale ; modified Berendsen thermostat tc-grps = Protein_HE1_CY2_ZN2_PRB Water_and_ions ; two coupling groups - more accurate tau_t = 0.1 0.1 ; time constant, in ps ref_t = 300 300 ; reference temperature, one for each group, in K ; Pressure coupling pcoupl = Parrinello-Rahman ; pressure coupling is on for NPT pcoupltype = isotropic ; uniform scaling of box vectors tau_p = 2.0 ; time constant, in ps ref_p = 1.0 ; reference pressure, in bar compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1 ; Periodic boundary conditions pbc = xyz ; 3-D PBC ; Dispersion correction DispCorr = EnerPres ; account for cut-off vdW scheme ; Velocity generation gen_vel = no ; assign velocities from Maxwell distribution
$ gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p 1PTR_ZAFF_PRB_solv_GMX.top -n index.ndx -o md_0_1.tpr
$ gmx mdrun -v -deffnm md_0_1