This note details the reproduction of the QM/MM modeling protocols described by Brás et al. (2018) using GROMACS (interfaced with CP2K).
Reference: Brás, N. F., et al., J. Phys. Chem. B 2018, 122, 3889–3899.doi: 10.1021/acs.jpcb.8b01321.
Contents
- 1 1. System Preparation
- 2 2. MD Simulation Protocol
- 3 2.1 Minimization and NVT Equilibration (303.15 K, 125 ps)
- 4 2.2 NPT Production with Triple Restraints (303.15 K, 1 bar, 10 ns)
- 5 2.3 NPT Production with Dual Restraints (10 ns)
- 6 2.4 Unrestrained NPT Run (10 ns)
- 7 2.5 Steered MD: Pulling along the Reaction Coordinate (0.2 ps)
1. System Preparation
Structural Assembly
Residue Recovery: The missing Q837 residue was extracted from the 3L4U.pdb structure and inserted into 2QMJ.pdb to generate 2QMJ_plus_Q837.pdb.
Ligand Alignment: A maltose structure was extracted from 8YBE.pdb and fitted to the acarbose molecule in 2QMJ.pdb to produce maltose_fitted.pdb. This file was processed via CHARMM-GUI’s Glycan Reader & Modeler to generate the GROMACS topology file (CARA.itp).
Assembly: After removing acarbose coordinates, the NEWPDB.PDB (generated by Antechamber) was inserted following the NAG glycan coordinates in 2QMJ.pdb, resulting in 2QMJ_maltose_input.pdb.
Q837_from_3L4U_aligned.pdb
ATOM 1 N GLN A 837 -56.319 -1.918 -54.152 1.00 52.24 N ATOM 2 CA GLN A 837 -56.489 -3.228 -53.471 1.00 51.57 C ATOM 3 C GLN A 837 -57.877 -3.508 -52.879 1.00 50.76 C ATOM 4 O GLN A 837 -58.136 -3.210 -51.705 1.00 51.36 O ATOM 5 CB GLN A 837 -55.377 -3.492 -52.436 1.00 51.89 C ATOM 6 CG GLN A 837 -55.042 -2.327 -51.488 1.00 52.50 C ATOM 7 CD GLN A 837 -55.174 -2.711 -50.028 1.00 52.02 C ATOM 8 OE1 GLN A 837 -54.177 -2.786 -49.310 1.00 49.07 O ATOM 9 NE2 GLN A 837 -56.420 -2.967 -49.583 1.00 50.20 N
maltose_fitted.pdb
HETATM 1 C1 GLC B 1 -18.637 -6.555 -5.479 1.00 20.00 C HETATM 2 C2 GLC B 1 -18.080 -5.440 -6.357 1.00 20.00 C HETATM 3 C3 GLC B 1 -18.902 -5.280 -7.625 1.00 20.00 C HETATM 4 C4 GLC B 1 -19.046 -6.614 -8.329 1.00 20.00 C HETATM 5 C5 GLC B 1 -19.495 -7.694 -7.358 1.00 20.00 C HETATM 6 C6 GLC B 1 -19.515 -9.049 -8.044 1.00 20.00 C HETATM 7 O1 GLC B 1 -19.972 -6.247 -5.064 1.00 20.00 O HETATM 8 O2 GLC B 1 -18.077 -4.209 -5.634 1.00 20.00 O HETATM 9 O3 GLC B 1 -18.244 -4.343 -8.478 1.00 20.00 O HETATM 10 O4 GLC B 1 -20.058 -6.525 -9.322 1.00 20.00 O HETATM 11 O5 GLC B 1 -18.621 -7.761 -6.237 1.00 20.00 O HETATM 12 O6 GLC B 1 -20.151 -9.974 -7.172 1.00 20.00 O HETATM 13 C1 GLC B 2 -19.555 -5.932 -10.525 1.00 19.82 C HETATM 14 C2 GLC B 2 -20.762 -5.492 -11.333 1.00 18.22 C HETATM 15 C3 GLC B 2 -21.625 -6.709 -11.603 1.00 19.03 C HETATM 16 C4 GLC B 2 -20.799 -7.724 -12.372 1.00 19.44 C HETATM 17 C5 GLC B 2 -19.558 -8.067 -11.566 1.00 24.91 C HETATM 18 C6 GLC B 2 -18.675 -9.055 -12.311 1.00 20.27 C HETATM 19 O2 GLC B 2 -21.513 -4.523 -10.609 1.00 26.01 O HETATM 20 O3 GLC B 2 -22.777 -6.339 -12.355 1.00 24.65 O HETATM 21 O4 GLC B 2 -21.561 -8.906 -12.618 1.00 28.65 O HETATM 22 O5 GLC B 2 -18.825 -6.875 -11.301 1.00 25.46 O HETATM 23 O6 GLC B 2 -17.756 -9.627 -11.384 1.00 20.51 O
CARA.itp
;;
;; Generated by CHARMM-GUI FF-Converter
;;
;; Correspondance:
;; jul316@lehigh.edu or wonpil@lehigh.edu
;;
;; GROMACS topology file for CARA
;;
[ moleculetype ]
; name nrexcl
CARA 3
[ atoms ]
; nr type resnr residu atom cgnr charge mass
1 CC3162 1 AGLC C1 1 0.340000 12.0110 ; qtot 0.340
2 HCA1 1 AGLC H1 2 0.090000 1.0080 ; qtot 0.430
3 OC311 1 AGLC O1 3 -0.650000 15.9994 ; qtot -0.220
4 HCP1 1 AGLC HO1 4 0.420000 1.0080 ; qtot 0.200
5 CC3163 1 AGLC C5 5 0.110000 12.0110 ; qtot 0.310
6 HCA1 1 AGLC H5 6 0.090000 1.0080 ; qtot 0.400
7 OC3C61 1 AGLC O5 7 -0.400000 15.9994 ; qtot 0.000
8 CC3161 1 AGLC C2 8 0.140000 12.0110 ; qtot 0.140
9 HCA1 1 AGLC H2 9 0.090000 1.0080 ; qtot 0.230
10 OC311 1 AGLC O2 10 -0.650000 15.9994 ; qtot -0.420
11 HCP1 1 AGLC HO2 11 0.420000 1.0080 ; qtot -0.000
12 CC3161 1 AGLC C3 12 0.140000 12.0110 ; qtot 0.140
13 HCA1 1 AGLC H3 13 0.090000 1.0080 ; qtot 0.230
14 OC311 1 AGLC O3 14 -0.650000 15.9994 ; qtot -0.420
15 HCP1 1 AGLC HO3 15 0.420000 1.0080 ; qtot -0.000
16 CC3161 1 AGLC C4 16 0.090000 12.0110 ; qtot 0.090
17 HCA1 1 AGLC H4 17 0.090000 1.0080 ; qtot 0.180
18 OC301 1 AGLC O4 18 -0.360000 15.9994 ; qtot -0.180
19 CC321 1 AGLC C6 19 0.050000 12.0110 ; qtot -0.130
20 HCA2 1 AGLC H61 20 0.090000 1.0080 ; qtot -0.040
21 HCA2 1 AGLC H62 21 0.090000 1.0080 ; qtot 0.050
22 OC311 1 AGLC O6 22 -0.650000 15.9994 ; qtot -0.600
23 HCP1 1 AGLC HO6 23 0.420000 1.0080 ; qtot -0.180
24 CC3162 2 AGLC C1 24 0.290000 12.0110 ; qtot 0.110
25 HCA1 2 AGLC H1 25 0.090000 1.0080 ; qtot 0.200
26 CC3163 2 AGLC C5 26 0.110000 12.0110 ; qtot 0.310
27 HCA1 2 AGLC H5 27 0.090000 1.0080 ; qtot 0.400
28 OC3C61 2 AGLC O5 28 -0.400000 15.9994 ; qtot -0.000
29 CC3161 2 AGLC C2 29 0.140000 12.0110 ; qtot 0.140
30 HCA1 2 AGLC H2 30 0.090000 1.0080 ; qtot 0.230
31 OC311 2 AGLC O2 31 -0.650000 15.9994 ; qtot -0.420
32 HCP1 2 AGLC HO2 32 0.420000 1.0080 ; qtot -0.000
33 CC3161 2 AGLC C3 33 0.140000 12.0110 ; qtot 0.140
34 HCA1 2 AGLC H3 34 0.090000 1.0080 ; qtot 0.230
35 OC311 2 AGLC O3 35 -0.650000 15.9994 ; qtot -0.420
36 HCP1 2 AGLC HO3 36 0.420000 1.0080 ; qtot -0.000
37 CC3161 2 AGLC C4 37 0.140000 12.0110 ; qtot 0.140
38 HCA1 2 AGLC H4 38 0.090000 1.0080 ; qtot 0.230
39 OC311 2 AGLC O4 39 -0.650000 15.9994 ; qtot -0.420
40 HCP1 2 AGLC HO4 40 0.420000 1.0080 ; qtot -0.000
41 CC321 2 AGLC C6 41 0.050000 12.0110 ; qtot 0.050
42 HCA2 2 AGLC H61 42 0.090000 1.0080 ; qtot 0.140
43 HCA2 2 AGLC H62 43 0.090000 1.0080 ; qtot 0.230
44 OC311 2 AGLC O6 44 -0.650000 15.9994 ; qtot -0.420
45 HCP1 2 AGLC HO6 45 0.420000 1.0080 ; qtot -0.000
[ bonds ]
; ai aj funct b0 Kb
1 2 1
1 3 1
1 7 1
1 8 1
3 4 1
5 6 1
5 7 1
16 5 1
5 19 1
8 9 1
8 10 1
8 12 1
10 11 1
12 13 1
12 14 1
12 16 1
14 15 1
16 17 1
16 18 1
18 24 1
19 20 1
19 21 1
19 22 1
22 23 1
24 25 1
24 28 1
24 29 1
26 27 1
26 28 1
37 26 1
26 41 1
29 30 1
29 31 1
29 33 1
31 32 1
33 34 1
33 35 1
33 37 1
35 36 1
37 38 1
37 39 1
39 40 1
41 42 1
41 43 1
41 44 1
44 45 1
[ pairs ]
; ai aj funct c6 c12 or
; ai aj funct fudgeQQ q1 q2 c6 c12
1 6 1
1 11 1
1 13 1
1 14 1
1 16 1
1 19 1
2 4 1
2 5 1
2 9 1
2 10 1
2 12 1
3 5 1
3 9 1
3 10 1
3 12 1
4 7 1
4 8 1
5 8 1
5 13 1
5 14 1
5 23 1
5 24 1
6 12 1
6 17 1
6 18 1
6 20 1
6 21 1
6 22 1
7 9 1
7 10 1
7 12 1
7 17 1
7 18 1
7 20 1
7 21 1
7 22 1
8 15 1
8 17 1
8 18 1
9 11 1
9 13 1
9 14 1
9 16 1
10 13 1
10 14 1
10 16 1
11 12 1
12 19 1
12 24 1
13 15 1
13 17 1
13 18 1
14 17 1
14 18 1
15 16 1
16 20 1
16 21 1
16 22 1
16 25 1
16 28 1
16 29 1
17 19 1
17 24 1
18 19 1
18 26 1
18 30 1
18 31 1
18 33 1
20 23 1
21 23 1
24 27 1
24 32 1
24 34 1
24 35 1
24 37 1
24 41 1
25 26 1
25 30 1
25 31 1
25 33 1
26 29 1
26 34 1
26 35 1
26 40 1
26 45 1
27 33 1
27 38 1
27 39 1
27 42 1
27 43 1
27 44 1
28 30 1
28 31 1
28 33 1
28 38 1
28 39 1
28 42 1
28 43 1
28 44 1
29 36 1
29 38 1
29 39 1
30 32 1
30 34 1
30 35 1
30 37 1
31 34 1
31 35 1
31 37 1
32 33 1
33 40 1
33 41 1
34 36 1
34 38 1
34 39 1
35 38 1
35 39 1
36 37 1
37 42 1
37 43 1
37 44 1
38 40 1
38 41 1
39 41 1
42 45 1
43 45 1
[ angles ]
; ai aj ak funct th0 cth S0 Kub
2 1 3 5
2 1 7 5
2 1 8 5
3 1 7 5
3 1 8 5
7 1 8 5
1 3 4 5
6 5 7 5
6 5 16 5
6 5 19 5
7 5 16 5
7 5 19 5
16 5 19 5
1 7 5 5
1 8 9 5
1 8 10 5
1 8 12 5
9 8 10 5
9 8 12 5
10 8 12 5
8 10 11 5
8 12 13 5
8 12 14 5
8 12 16 5
13 12 14 5
13 12 16 5
14 12 16 5
12 14 15 5
5 16 12 5
5 16 17 5
5 16 18 5
12 16 17 5
12 16 18 5
17 16 18 5
16 18 24 5
5 19 20 5
5 19 21 5
5 19 22 5
20 19 21 5
20 19 22 5
21 19 22 5
19 22 23 5
18 24 25 5
18 24 28 5
18 24 29 5
25 24 28 5
25 24 29 5
28 24 29 5
27 26 28 5
27 26 37 5
27 26 41 5
28 26 37 5
28 26 41 5
37 26 41 5
24 28 26 5
24 29 30 5
24 29 31 5
24 29 33 5
30 29 31 5
30 29 33 5
31 29 33 5
29 31 32 5
29 33 34 5
29 33 35 5
29 33 37 5
34 33 35 5
34 33 37 5
35 33 37 5
33 35 36 5
26 37 33 5
26 37 38 5
26 37 39 5
33 37 38 5
33 37 39 5
38 37 39 5
37 39 40 5
26 41 42 5
26 41 43 5
26 41 44 5
42 41 43 5
42 41 44 5
43 41 44 5
41 44 45 5
[ dihedrals ]
; ai aj ak al funct phi0 cp mult
2 1 3 4 9
2 1 7 5 9
3 1 7 5 9
2 1 8 9 9
2 1 8 10 9
2 1 8 12 9
3 1 8 9 9
3 1 8 10 9
3 1 8 12 9
7 1 8 9 9
7 1 8 10 9
7 1 8 12 9
4 3 1 7 9
4 3 1 8 9
6 5 16 12 9
6 5 16 17 9
6 5 16 18 9
7 5 16 12 9
7 5 16 17 9
7 5 16 18 9
6 5 19 20 9
6 5 19 21 9
6 5 19 22 9
7 5 19 20 9
7 5 19 21 9
7 5 19 22 9
16 5 19 20 9
16 5 19 21 9
16 5 19 22 9
5 7 1 8 9
1 7 5 6 9
1 7 5 16 9
1 7 5 19 9
1 8 10 11 9
9 8 10 11 9
1 8 12 13 9
1 8 12 14 9
1 8 12 16 9
9 8 12 13 9
9 8 12 14 9
9 8 12 16 9
10 8 12 13 9
10 8 12 14 9
10 8 12 16 9
11 10 8 12 9
8 12 14 15 9
13 12 14 15 9
8 12 16 17 9
8 12 16 18 9
13 12 16 17 9
13 12 16 18 9
14 12 16 17 9
14 12 16 18 9
15 14 12 16 9
12 16 5 19 9
17 16 5 19 9
18 16 5 19 9
5 16 12 8 9
5 16 12 13 9
5 16 12 14 9
5 16 18 24 9
12 16 18 24 9
17 16 18 24 9
16 18 24 25 9
16 18 24 28 9
16 18 24 29 9
5 19 22 23 9
20 19 22 23 9
21 19 22 23 9
18 24 28 26 9
25 24 28 26 9
18 24 29 30 9
18 24 29 31 9
18 24 29 33 9
25 24 29 30 9
25 24 29 31 9
25 24 29 33 9
28 24 29 30 9
28 24 29 31 9
28 24 29 33 9
27 26 37 33 9
27 26 37 38 9
27 26 37 39 9
28 26 37 33 9
28 26 37 38 9
28 26 37 39 9
27 26 41 42 9
27 26 41 43 9
27 26 41 44 9
28 26 41 42 9
28 26 41 43 9
28 26 41 44 9
37 26 41 42 9
37 26 41 43 9
37 26 41 44 9
26 28 24 29 9
24 28 26 27 9
24 28 26 37 9
24 28 26 41 9
24 29 31 32 9
30 29 31 32 9
24 29 33 34 9
24 29 33 35 9
24 29 33 37 9
30 29 33 34 9
30 29 33 35 9
30 29 33 37 9
31 29 33 34 9
31 29 33 35 9
31 29 33 37 9
32 31 29 33 9
29 33 35 36 9
34 33 35 36 9
29 33 37 38 9
29 33 37 39 9
34 33 37 38 9
34 33 37 39 9
35 33 37 38 9
35 33 37 39 9
36 35 33 37 9
33 37 26 41 9
38 37 26 41 9
39 37 26 41 9
26 37 33 29 9
26 37 33 34 9
26 37 33 35 9
26 37 39 40 9
33 37 39 40 9
38 37 39 40 9
26 41 44 45 9
42 41 44 45 9
43 41 44 45 9
#ifdef POSRES
[ position_restraints ]
1 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
3 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
5 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
7 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
8 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
10 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
12 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
14 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
16 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
18 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
19 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
22 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
24 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
26 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
28 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
29 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
31 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
33 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
35 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
37 1 POSRES_FC_BB POSRES_FC_BB POSRES_FC_BB
39 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
41 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
44 1 POSRES_FC_SC POSRES_FC_SC POSRES_FC_SC
#endif
#ifdef DIHRES
[ dihedral_restraints ]
1 7 5 16 1 60.0 2.5 DIHRES_FC
7 5 16 12 1 -60.0 2.5 DIHRES_FC
5 16 12 8 1 60.0 2.5 DIHRES_FC
16 12 8 1 1 -60.0 2.5 DIHRES_FC
12 8 1 7 1 60.0 2.5 DIHRES_FC
8 1 7 5 1 -60.0 2.5 DIHRES_FC
24 28 26 37 1 60.0 2.5 DIHRES_FC
28 26 37 33 1 -60.0 2.5 DIHRES_FC
26 37 33 29 1 60.0 2.5 DIHRES_FC
37 33 29 24 1 -60.0 2.5 DIHRES_FC
33 29 24 28 1 60.0 2.5 DIHRES_FC
29 24 28 26 1 -60.0 2.5 DIHRES_FC
#endif
obabel -ipdb maltose_fitted.pdb -h -opdb -O maltose_fitted_h.pdb antechamber -fi pdb -i maltose_fitted_h.pdb -fo prepi -o maltose.prep obabel -ipdb NEWPDB.PDB -omol2 -O maltose_antechamber.mol2 sed -i "" "s/GLC /GLC B/g" NEWPDB.PDB
CHARMM-GUI Workflow
The 2QMJ_maltose_input.pdb was processed through the following steps:
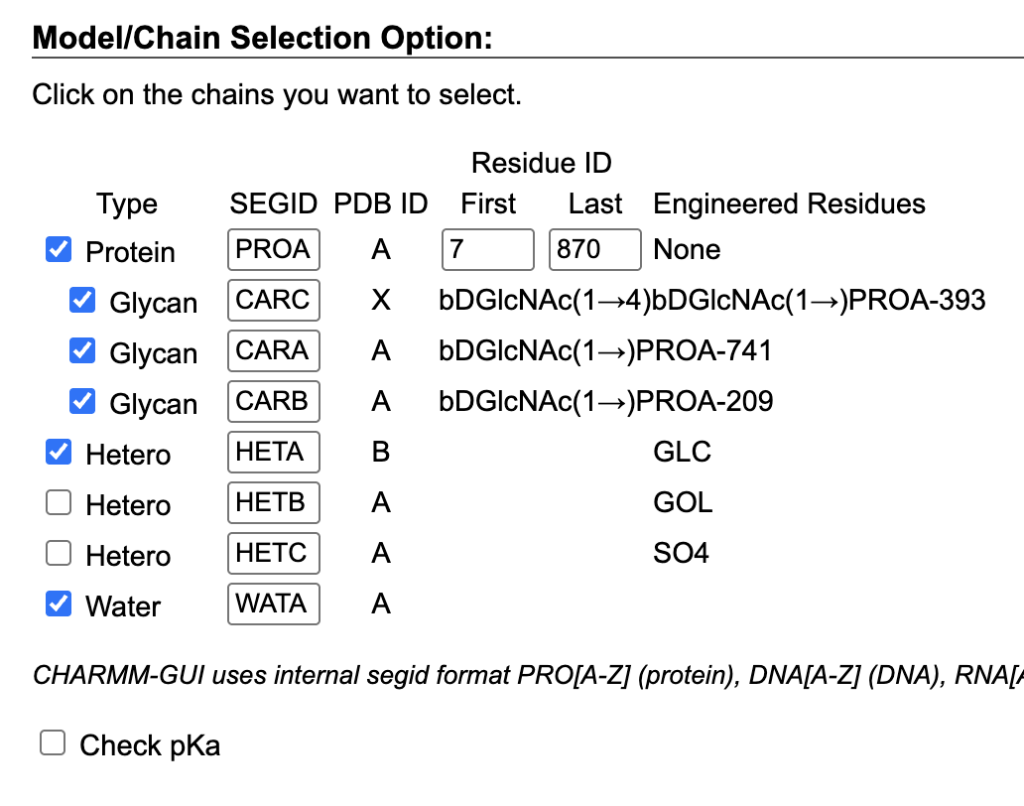
- Input Generator > Glycan Reader & Modeler: Uploaded the PDB file.
- Component Selection: Selected Protein, HETA, and Water.
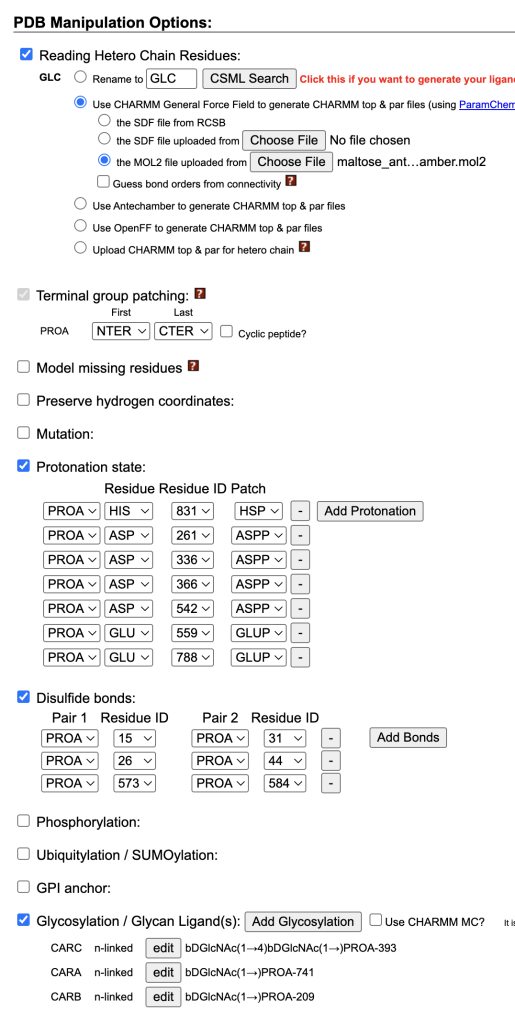
- Ligand Parameters: Set “Reading Hetero Chain Residues” to use CHARMM General Force Field (CGenFF).
- Uploaded maltose_antechamber.mol2.
- Unchecked “Model missing residues.”
- Manually set protonation states for: HIS831, ASP261, ASP336, ASP366, ASP542, GLU559, and GLU788.
- Solvation & Ion addition: Set the edge distance to 12.0 Å. Added 150 mM NaCl (standardizing the ionic strength rather than only adding 23 Na⁺ ions to neutralize).
Topology Finalization
The CGenFF-generated maltose topology and coordinates were manually replaced with the outputs from the Glycan Reader & Modeler to create step3_input_modified.pdb. In topol.top, the downloaded CHARMM36 force field was included, and the “HETA” residue name was updated to “CARA.”
step3_input_modified.pdb
ATOM 13624 H62 BGLC 1 71.007 48.075 80.029 1.00 0.00 CARB H ATOM 13625 O6 BGLC 1 72.397 49.312 79.163 1.00 0.00 CARB O ATOM 13626 HO6 BGLC 1 72.514 49.786 78.333 1.00 0.00 CARB H TER ATOM 13627 C1 AGLC 1 64.666 41.110 70.306 1.00 0.00 CARA C ATOM 13628 H1 AGLC 1 65.508 40.620 70.843 1.00 0.00 CARA H ATOM 13629 O1 AGLC 1 64.261 42.275 71.033 1.00 0.00 CARA O ATOM 13630 HO1 AGLC 1 63.972 41.976 71.903 1.00 0.00 CARA H ATOM 13631 C5 AGLC 1 62.444 40.803 69.577 1.00 0.00 CARA C ATOM 13632 H5 AGLC 1 62.104 41.710 70.140 1.00 0.00 CARA H ATOM 13633 O5 AGLC 1 63.579 40.198 70.187 1.00 0.00 CARA O ATOM 13634 C2 AGLC 1 65.120 41.488 68.900 1.00 0.00 CARA C ATOM 13635 H2 AGLC 1 65.461 40.556 68.391 1.00 0.00 CARA H ATOM 13636 O2 AGLC 1 66.213 42.403 68.974 1.00 0.00 CARA O ATOM 13637 HO2 AGLC 1 65.972 43.129 69.565 1.00 0.00 CARA H ATOM 13638 C3 AGLC 1 63.984 42.118 68.111 1.00 0.00 CARA C ATOM 13639 H3 AGLC 1 63.726 43.108 68.553 1.00 0.00 CARA H ATOM 13640 O3 AGLC 1 64.416 42.299 66.762 1.00 0.00 CARA O ATOM 13641 HO3 AGLC 1 65.340 41.994 66.681 1.00 0.00 CARA H ATOM 13642 C4 AGLC 1 62.760 41.227 68.152 1.00 0.00 CARA C ATOM 13643 H4 AGLC 1 62.925 40.308 67.542 1.00 0.00 CARA H ATOM 13644 O4 AGLC 1 61.627 41.949 67.690 1.00 0.00 CARA O ATOM 13645 C6 AGLC 1 61.293 39.812 69.593 1.00 0.00 CARA C ATOM 13646 H61 AGLC 1 61.544 38.851 69.092 1.00 0.00 CARA H ATOM 13647 H62 AGLC 1 60.407 40.284 69.159 1.00 0.00 CARA H ATOM 13648 O6 AGLC 1 60.893 39.625 70.945 1.00 0.00 CARA O ATOM 13649 HO6 AGLC 1 61.722 39.574 71.442 1.00 0.00 CARA H ATOM 13650 C1 AGLC 2 61.574 41.975 66.260 1.00 0.00 CARA C ATOM 13651 H1 AGLC 2 62.495 42.191 65.705 1.00 0.00 CARA H ATOM 13652 C5 AGLC 2 59.800 40.395 66.308 1.00 0.00 CARA C ATOM 13653 H5 AGLC 2 59.842 40.325 67.393 1.00 0.00 CARA H ATOM 13654 O5 AGLC 2 61.055 40.757 65.739 1.00 0.00 CARA O ATOM 13655 C2 AGLC 2 60.621 43.093 65.877 1.00 0.00 CARA C ATOM 13656 H2 AGLC 2 60.526 43.143 64.768 1.00 0.00 CARA H ATOM 13657 O2 AGLC 2 61.111 44.342 66.353 1.00 0.00 CARA O ATOM 13658 HO2 AGLC 2 61.356 44.301 67.297 1.00 0.00 CARA H ATOM 13659 C3 AGLC 2 59.270 42.804 66.503 1.00 0.00 CARA C ATOM 13660 H3 AGLC 2 59.357 42.759 67.616 1.00 0.00 CARA H ATOM 13661 O3 AGLC 2 58.345 43.836 66.175 1.00 0.00 CARA O ATOM 13662 HO3 AGLC 2 58.631 44.627 66.649 1.00 0.00 CARA H ATOM 13663 C4 AGLC 2 58.774 41.468 65.981 1.00 0.00 CARA C ATOM 13664 H4 AGLC 2 58.641 41.524 64.876 1.00 0.00 CARA H ATOM 13665 O4 AGLC 2 57.518 41.134 66.572 1.00 0.00 CARA O ATOM 13666 HO4 AGLC 2 57.343 41.641 67.380 1.00 0.00 CARA H ATOM 13667 C6 AGLC 2 59.367 39.040 65.772 1.00 0.00 CARA C ATOM 13668 H61 AGLC 2 59.501 38.989 64.669 1.00 0.00 CARA H ATOM 13669 H62 AGLC 2 58.297 38.845 66.005 1.00 0.00 CARA H ATOM 13670 O6 AGLC 2 60.140 38.032 66.418 1.00 0.00 CARA O ATOM 13671 HO6 AGLC 2 61.044 38.025 66.070 1.00 0.00 CARA H TER ATOM 13672 SOD SOD 1 47.130 96.053 99.155 1.00 0.00 IONSNa ATOM 13673 SOD SOD 2 77.410 92.297 2.916 1.00 0.00 IONSNa ATOM 13674 SOD SOD 3 41.610 89.017 71.986 1.00 0.00 IONSNa
topol.top (include downloaded charmm36 force field and replace “HETA” with “CARA”)
; Include forcefield parameters #include "charmm36-feb2026_cgenff-5.0.ff/forcefield.itp" ; modified #include "toppar/PROA.itp" #include "toppar/CARA.itp" ; modified #include "toppar/SOD.itp" #include "toppar/TIP3.itp" [ system ] ; Name Title [ molecules ] ; Compound #mols PROA 1 CARA 1 ; modified SOD 23 TIP3 42746
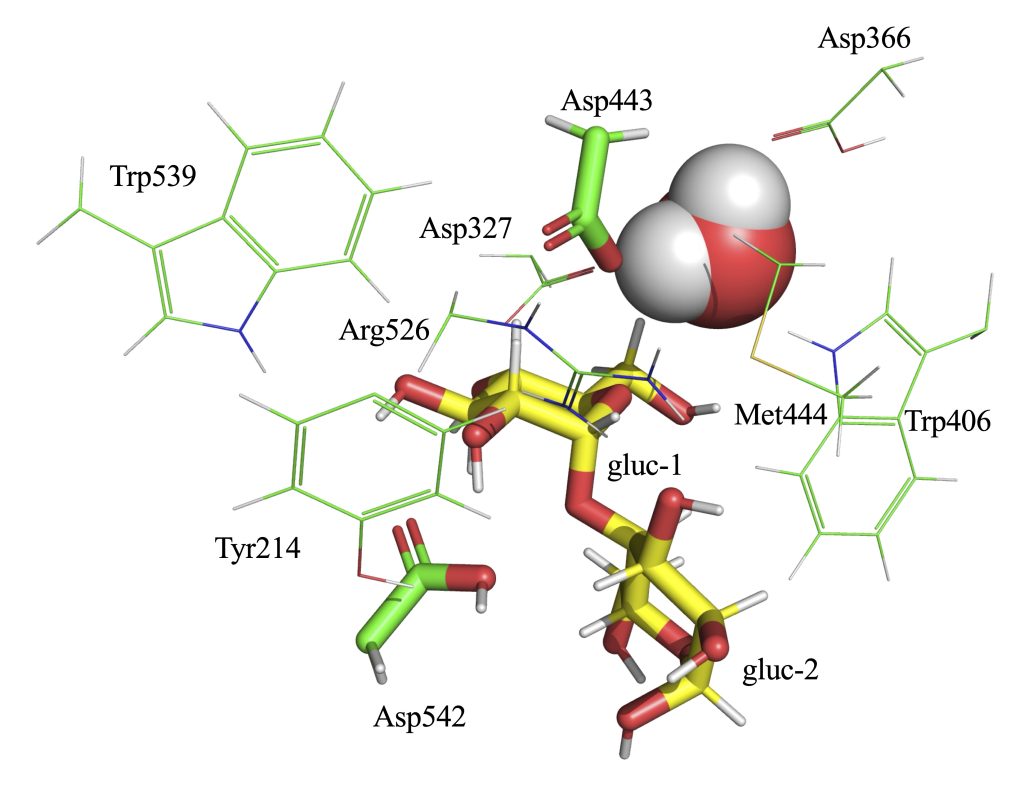
cp -r charmm-gui-[numbers]/gromacs . cd gromacs source /usr/local/gromacs/2026.1_cuda_torchcu128/bin/GMXRC gmx editconf -f step3_input_modified.pdb -bt cubic -box 11.7 -o box.gro gmx make_ndx -f box.gro <<EOF 1 | 13 | 14 15 | 16 | 17 name 18 SOLU name 19 SOLV a 3242-3253 name 20 TYR214 a 5010-5015 name 21 ASP327 a 5647-5653 name 22 ASPP366 a 6223-6240 name 23 TRP406 a 6828-6833 name 24 ASP443 a 6843-6850 name 25 MET444 a 8100-8111 name 26 ARG526 a 8277-8294 name 27 TRP539 a 8327-8333 name 28 ASPP542 a 14190-14192 name 29 HOH 14|20|21|22|23|24|25|26|27|28|29 name 30 QMatoms a 6833 a 8333 a 13644 a 13650 name 31 ASP443_OD2 name 32 ASP542_HD2 name 33 AGL1_O4 name 34 AGL2_C1 q EOF
2. MD Simulation Protocol
2.1 Minimization and NVT Equilibration (303.15 K, 125 ps)
To maintain a complex geometry similar to the acarbose-bound state, flat-bottom distance restraints and angle restraints were applied using the GROMACS pull code (pull rate = 0):
Angle: ASP443_OD2 – AGL2_C1 – AGL1_O4 (nucleophile–anomeric carbon–leaving atom)
Distance 1: ASP443_OD2 – AGL2_C1
Distance 2: AGL1_O4 – ASP542_HD2
; PULL CODE settings pull = yes pull-ngroups = 4 pull-ncoords = 3 ; Define the groups by their names in the index file pull-group1-name = ASP443_OD2 pull-group2-name = AGL2_C1 pull-group3-name = AGL1_O4 pull-group4-name = ASP542_HD2 ; --- Pull Coordinate 1 (Atom 1 and Atom 2) --- pull-coord1-groups = 1 2 ; References pull-group1 and pull-group2 pull-coord1-type = flat-bottom ; Or 'constant-force', 'constraint', etc. pull-coord1-geometry = distance ; Pulling along the vector connecting them pull-coord1-dim = Y Y Y ; Pulling in all dimensions (3D distance) pull-coord1-init = 0.33 ; Use initial distance as reference pull-coord1-k = 1000 ; Force constant [kJ/(mol*nm^2)] pull-coord1-rate = 0 ; Pulling rate [nm/ps] ; --- Pull Coordinate 2 (Atom 1, Atom 2, and Atom 3) --- pull-coord2-groups = 2 1 2 3 ; vector 2-1 and vector 2-3 pull-coord2-type = umbrella ; Applies a harmonic restraint pull-coord2-geometry = angle ; Specifies an angle (deg) pull-coord2-dim = Y Y Y ; Dimensions are ignored for angle pull-coord2-start = no ; Do not use the initial MD angle as reference pull-coord2-init = 180.0 ; The target angle in degrees pull-coord2-k = 500.0 ; Force constant (kJ/mol/rad^2) ; --- Pull Coordinate 3 (Atom 3 and Atom 4) --- pull-coord3-groups = 3 4 ; References pull-group3 and pull-group4 pull-coord3-type = flat-bottom pull-coord3-geometry = distance pull-coord3-dim = Y Y Y pull-coord3-init = 0.2 pull-coord3-k = 1000 pull-coord3-rate = 0
wget -O charmm36-feb2026_cgenff-5.0.ff.tgz https://mackerell.umaryland.edu/download.php?filename=CHARMM_ff_params_files/charmm36-feb2026_cgenff-5.0.ff.tgz tar xvzf charmm36-feb2026_cgenff-5.0.ff.tgz gmx grompp -f step4.0_minimization_pull.mdp -c box.gro -r box.gro -n index.ndx -o step4.0 gmx mdrun -v -deffnm step4.0 -ntmpi 1 -ntomp 9 gmx grompp -f step4.1_equilibration_pull.mdp -c step4.0.gro -r step4.0.gro -n index.ndx -o step4.1 gmx mdrun -v -deffnm step4.1 -ntmpi 1 -ntomp 9 # 125 ps gmx grompp -f step5_production_pull.mdp -c step4.1.gro -t step4.1.cpt -n index.ndx -o step5_1 gmx mdrun -v -deffnm step5_1 -ntmpi 1 -ntomp 9 # 10 ns In PyMOL, select near_contacts, byres ((polymer.protein or resn TIP3) within 5 of resn AGLC) In VMD, (protein or resname TIP3) and same residue as within 5 of resname AGLC
run_equilibration.sh
#!/bin/bash #SBATCH --job-name=step5_1_2_3 #SBATCH --output=job_%j.out #SBATCH --error=job_%j.err #SBATCH --partition=main #SBATCH --nodes=1 #SBATCH --ntasks=1 #SBATCH --cpus-per-task=9 # number of CPU core #SBATCH --mem=2G # memory #SBATCH --gres=gpu:1 # #SBATCH --time=48:00:00 # max job time cd $SLURM_SUBMIT_DIR source /usr/local/gromacs/2026.1_cuda_torchcu128/bin/GMXRC OMP=9 gmx grompp -f step5_production_pull.mdp -c step4.1.gro -t step4.1.cpt -n index.ndx -o step5_1 gmx mdrun -deffnm step5_1 -ntmpi 1 -ntomp 9 # with three restraints, 10 ns gmx grompp -f step5_production_pull_2.mdp -c step5_1.gro -t step5_1.cpt -n index.ndx -o step5_2 gmx mdrun -deffnm step5_2 -ntmpi 1 -ntomp 9 # with two restraints between ASP443 and maltose, 10 ns gmx grompp -f step5_production_10ns.mdp -c step5_2.gro -t step5_2.cpt -n index.ndx -o step5_3 gmx mdrun -deffnm step5_3 -ntmpi 1 -ntomp 9 # without restraint, 10 ns
sudo systemctl start slurmctld sudo systemctl enable slurmctld sudo systemctl start slurmd sudo systemctl enable slurmd sudo scontrol update nodename=mas-i9wx state=resume sbatch run_equilibration.sh
2.2 NPT Production with Triple Restraints (303.15 K, 1 bar, 10 ns)
The system was stabilized with all three restraints active.
gmx grompp -f step5_production_pull.mdp -c step4.1.gro -t step4.1.cpt -n index.ndx -o step5_1 gmx mdrun -v -deffnm step5_1 -ntmpi 1 -ntomp 9 # with three restraints, 10 ns
2.3 NPT Production with Dual Restraints (10 ns)
The restraint on the ASP542_HD2 – AGL1_O4 distance (Coordinate 3) was removed.
Observation: Without this restraint, the ASP542 side chain reoriented away from the maltose.
gmx grompp -f step5_production_pull_2.mdp -c step5_1.gro -t step5_1.cpt -n index.ndx -o step5_2 gmx mdrun -v -deffnm step10_2 -ntmpi 1 -ntomp 9 # 10 ns
2.4 Unrestrained NPT Run (10 ns)
A fully unrestrained simulation was performed.
Correction: Consequently, the coordinates from the NPT simulation with three restraints (Section 2.2) were used as the starting point for the subsequent pulling simulations.
Result: Similar to section 2.3, the ASP542 side chain maintained an incorrect orientation relative to the substrate.
gmx grompp -f step5_production_10ns.mdp -c step5_2.gro -t step5_2.cpt -n index.ndx -o step5_3 gmx mdrun -v -deffnm step5_3 -ntmpi 1 -ntomp 9 # 10 ns
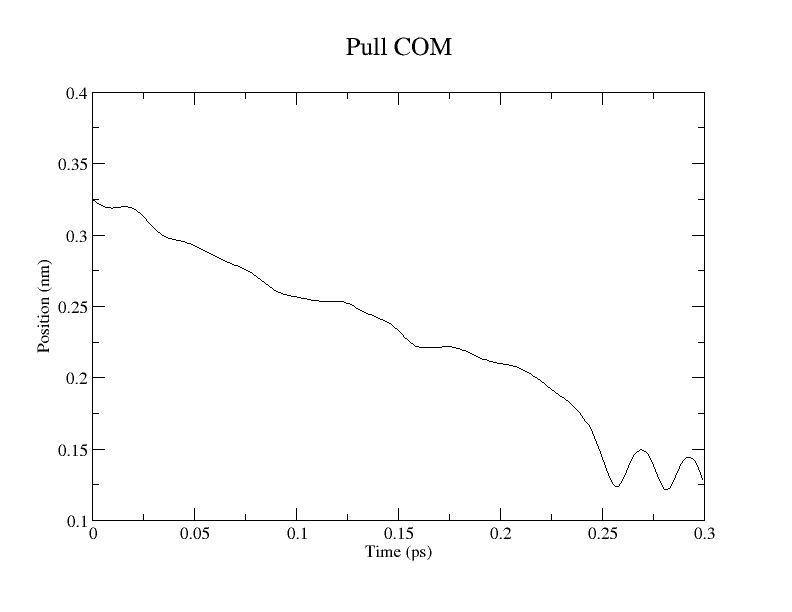
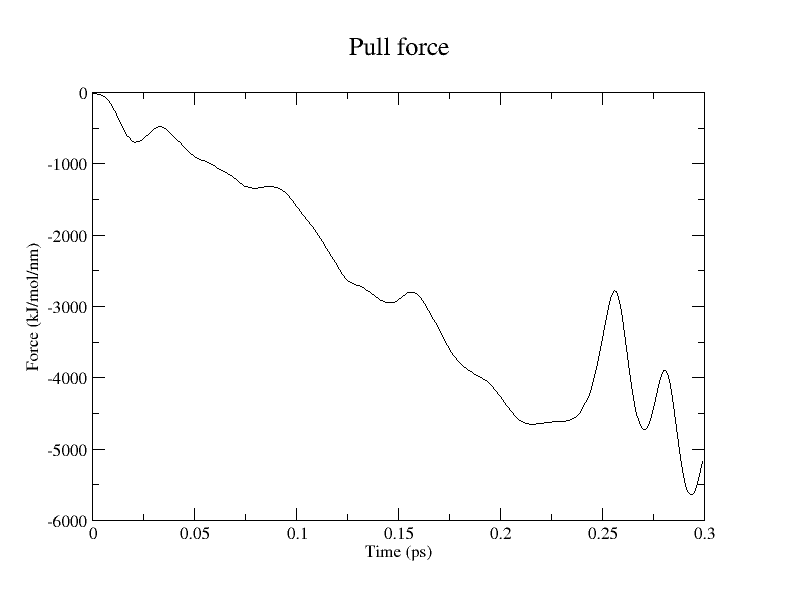
2.5 Steered MD: Pulling along the Reaction Coordinate (0.2 ps)
Due to memory limitations encountered when using GROMACS 2026.1 with CP2K 2026.1 on a local workstation, subsequent steered MD (SMD) simulations were performed using GROMACS 2025.4 and CP2K 2024.3 at the Research Center for Computational Science (Okazaki, Japan). This version compatibility necessitated restricting the DFT functionals to PBE/TZVP-MOLOPT and BLYP/TZVP-MOLOPT.
We performed pulling along the ASP443_OD2 – AGL2_C1 coordinate to model the first glycosylation step. pull-coord1-k, 50000 [kJ/(mol*nm^2)]; pull-coord1-rate, -1.0 [nm/ps]. Per Brás et al. (2018), the target distances for this step are:
Reactant (R): 3.26 Å
Transition State (TS1): 2.05 Å
Intermediate (INT1): 1.49 Å
step6_qmmm_pulling.mdp
integrator = md dt = 0.001 nsteps = 400 nstxout-compressed = 1 nstxout = 0 nstvout = 0 nstfout = 0 nstcalcenergy = 1 nstenergy = 1 nstlog = 1 ; cutoff-scheme = Verlet nstlist = 20 vdwtype = Cut-off vdw-modifier = Force-switch rvdw_switch = 1.0 rvdw = 1.2 rlist = 1.2 rcoulomb = 1.2 coulombtype = PME ; tcoupl = v-rescale tc_grps = SOLU SOLV tau_t = 1.0 1.0 ref_t = 303.15 303.15 ; ; constraints = none constraint_algorithm = LINCS continuation = yes ; nstcomm = 100 comm_mode = linear comm_grps = SOLU SOLV ; ; CP2K QMMM parameters qmmm-cp2k-active = true ; Activate QMMM MdModule qmmm-cp2k-qmgroup = QMatoms ; Index group of QM atoms qmmm-cp2k-qmmethod = PBE ; Method to use qmmm-cp2k-qmcharge = -1 ; Charge of QM system qmmm-cp2k-qmmultiplicity = 1 ; Multiplicity of QM system ; PULL CODE settings pull = yes pull-ngroups = 2 pull-ncoords = 1 ; Define the groups by their names in the index file pull-group1-name = ASP443_OD2 pull-group2-name = AGL2_C1 ; --- Pull Coordinate 1 (Atom 1 and Atom 2) --- pull-coord1-groups = 1 2 ; References pull-group1 and pull-group2 pull-coord1-type = umbrella ; Or 'constant-force', 'constraint', etc. pull-coord1-geometry = distance ; Pulling along the vector connecting them pull-coord1-dim = Y Y Y ; Pulling in all dimensions (3D distance) pull-coord1-start = yes pull-coord1-k = 50000 ; Force constant [kJ/(mol*nm^2)] pull-coord1-rate = -1.0 ; Pulling rate [nm/ps] pull-nstxout = 1 pull-nstfout = 1
Execution and Performance
The simulation was executed using the following commands:
source /usr/local/gromacs/2026.1_mpi_d_cp2k/bin/GMXRC gmx_mpi_d_cp2k grompp -f step6_qmmm_pulling_test.mdp -c step5_1.gro -t step5_1.cpt -n index.ndx -o step6_qmmm_test mpirun -np 32 gmx_mpi_d_cp2k mdrun -v -deffnm step6_qmmm_test -ntomp 1 # 5 fs (5 steps)
The calculation required 6.5 hours to complete 300 steps (0.3 ps) using 32 CPU cores.
Conclusion and Visualization
While QM/MM MD simulations provide a reliable framework for estimating free energy changes, their computational cost remains high. Consequently, static QM/MM methods employing the ONIOM scheme remain a practical alternative. Snapshots extracted from this pulling simulation can serve as initial coordinates for subsequent ONIOM calculations.
This blog post was copyedited with the assistance of Gemini (Google LLC). The author has reviewed and verified the final content for accuracy.