環境: macOS Sierra 10.12.1, 3.3 GHz Intel Core i5, メモリ8 GB.AmberTools 16, Gromacs 5.1.4.
参考: An Amber Lipid Force Field Tutorial: Lipid14 Edition
この記事は主に自分と研究室の学生向けのメモです.C1ドメインはリガンドが結合することで,膜へトランスロケーションし,膜とリガンド,タンパク質の複合体を作ります (peripheral membrane protein).C1ドメインはphosphatidylserine (PS) に対して特異性を持ちますが,AmberのLipid14力場は,NPγT(温度・圧力・表面張力一定)やNPAT(温度・圧力・膜面積一定)条件が必要だったLipid11力場とは異なり,NPT(温度・圧力一定)条件で膜の面積が維持されるようになりました.しかし,Lipid14は現在phosphatidylcholine (PC) とphosphatidylethanolamine (PE) のパラメータしか持たないため,リン脂質にはPOPCを選択しました.Amber Lipid11力場やcharmm力場にはPSのパラメータが含まれています.
タンパク質構造にはPKCδ C1Bドメインとphorbol 13-acetateの複合体結晶構造 (1PTR.pdb) を使用します.
Contents
CHARMM-GUIに入力するファイルの準備
タンパク質構造の修正については別記事を参考にしてください (modeller.pdbとする).
PPM Serverでは膜タンパク質や膜近傍タンパク質の膜中の配向や位置を計算してくれます.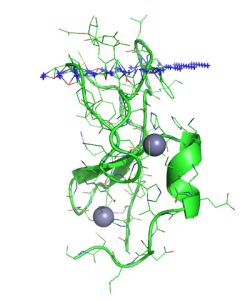 Include heteroatoms, excluding water and detergents, for positioning in membrane: yesにチェックを入れて,upload your PDB fileからファイルを選択し,submitボタンを押します.数秒で結果が出ます.膜へ挿入される長さ,角度,膜挿入の自由エネルギー変化などが書かれています.Download Output Fileから出力されたPDBファイルをダウンロードします (modellerout.pdb).青いダミー原子はリン脂質のカルボニル基の位置に相当します.
Include heteroatoms, excluding water and detergents, for positioning in membrane: yesにチェックを入れて,upload your PDB fileからファイルを選択し,submitボタンを押します.数秒で結果が出ます.膜へ挿入される長さ,角度,膜挿入の自由エネルギー変化などが書かれています.Download Output Fileから出力されたPDBファイルをダウンロードします (modellerout.pdb).青いダミー原子はリン脂質のカルボニル基の位置に相当します.
出力ファイルには水素原子が含まれていないので,リガンドの座標を抽出してPRB.pdbとして,Avogadroで水素原子を付加します.Avogadroで付加した水素原子の先頭はATOM,残基番号は1,残基名はLIGになっているので,それぞれ,HETATM,55,PRBに統一しました.Antechamberを使ってそれぞれの原子に固有のIDを振り,babelでmol2形式のファイルを作ります (CHARMM-GUIへの入力に必要).
$ antechamber -fi pdb -fo pdb -i PRB.pdb -o PRB_AC.pdb
$ babel PRB_AC.pdb PRB_AC.mol2PRB_ante.pdbの先頭をATOMからHETATMへ変更し,modellerout.pdbのリガンド座標に上書きして保存します.また,亜鉛に配位するCYS残基の残基名をCYMに (電荷を合わせるため),HIS残基 (HIS2, HIS40) の残基名をHSEに (水素原子の位置を指定するため) 変更します.修正したPDBファイルの名称はmodellerout_mod.pdbとします.
CHARMM-GUIによる膜-リガンド-タンパク質複合体構造の構築
CHARMM-GUIのMembrane Builderから修正したPDBファイル (modellerout_mod.pdb) をアップロードします.
- ファイルを選択 > modellerout_mod.pdb (修正済) > PDBにチェック > Next Step
- Model/Chain Selection Option > ZNとPRBにチェック > Next Step
- Renaming Engineered Residues: Rename CYM to CYM
- PRB > Use CHARMM General Force Field to generate CHARMM top & par files > the MOL2 file uploaded from にチェックしてPRB_AC.mol2を選択.下の方のHas three membered ringにチェック > Next Step
- Orientations Options: Use PDB Orientation > Next Step
- System Size Determination Options: Homogeneous Lipid > Use Geometry > Length of X: 66 (Upper leafletに含まれるリン脂質が64分子になる長さ), Lipid Type: POPC > Next Step > 全てデフォルトのままでNext Step > Next Step (少々時間がかかる) > Next Step
- System SizeのA, B, Cの値をメモしておく > Next Step (少々時間がかかる)
- download .tgzをクリックしてファイルをダウンロード
解凍したファイルに含まれるstep5_assembly.pdbを以後使用します.
AmberToolsによるトポロジーの生成
CHARMMの脂質は1つの残基になっていますが,Amber Lipid14力場ではモジュール構造になっています.Amberが読み込める形式にするため,AmberTools16付属のスクリプトを使います.
$ charmmlipid2amber.py -i step5_assembly.pdb -c $AMBERHOME/dat/charmmlipid2amber/charmmlipid2amber.csv -o 1PTR_POPC.pdbtleapでの処理のために,
- タンパク質の水素原子を除去 ($ babel -d protein.pdb protein_noh.pdbとタンパク質部分のみ処理してから元ファイルに上書きする).GLY52のOT2原子の行を削除.GLY52のOT1原子をOに修正.
- 残基名をCYM→CY2,HSE2とHSE40→HE1,HSD→HIDに修正.タンパク質とZNの間,ZNとZNの間,ZNとリガンドの間,リガンド (PRB) と脂質 (PA) の間にTER行を挟みます.
- 1PTR_POPC_ZAFF.pdbとして保存.
修正後のファイル (1PTR_POPC_ZAFF.pdb) の一部分:
ATOM 408 C GLY 52 -0.993 -8.981 -39.022 1.00 0.00 C ATOM 409 O GLY 52 -1.727 -8.677 -38.044 1.00 0.00 O TER ATOM 804 ZN ZN2 53 -1.831 -3.757 -34.565 1.00 0.00 HETA TER ATOM 805 ZN ZN2 54 7.207 2.614 -24.450 1.00 0.00 HETA TER ATOM 806 C8 PRB 55 -1.115 -2.860 -12.178 1.00 0.00 HETB ATOM 807 C9 PRB 55 0.100 -3.129 -11.198 1.00 0.00 HETB ATOM 808 H8 PRB 55 -1.358 -1.776 -12.164 1.00 0.00 HETB
亜鉛ファインガーのためのPREPファイルとfrcmodファイルを作業ディレクトリにコピーします.
$ cp $AMBERHOME/dat/mtkpp/ZAFF/201108/ZAFF.prep $AMBERHOME/dat/mtkpp/ZAFF/201108/ZAFF.frcmod .リガンドファイルのAmber用トポロジーを作成します.AM1-BCC電荷 (-c bcc) は,非経験的分子軌道計算によるRESP (Restrained ElectroStatic Potential) 電荷に近い値を半経験的手法により計算した電荷です.MM法では原子核の位置に部分電荷を置きますが,本来の電子雲の分布による分子の静電ポテンシャルをこの部分電荷が再現できるように値を決める必要があります.
$ antechamber -fi pdb -i PRB_AC.pdb -fo prepi -o PRB.prep -c bcc -at gaff2
$ parmchk2 -i PRB.prep -o PRB.frcmod -f prepi -s gaff2以下のスクリプトをtleapで実行します.
tleap.in
source leaprc.protein.ff14SB #Source the ff14SB force field
source leaprc.water.tip3p #Source water parameters
source leaprc.lipid14 #Source the Lipid14 force field
source leaprc.gaff2 #Source the GAFF2 force filed for small molecules
addAtomTypes { { "ZN" "Zn" "sp3" } { "S2" "S" "sp3" } { "N1" "N" "sp3" } } #Add atom types for the ZAFF metal center with Center ID 2
loadamberprep ZAFF.prep #Load ZAFF prep file
loadamberparams ZAFF.frcmod #Load ZAFF frcmod file
loadamberprep PRB.prep
loadamberparams PRB.frcmod
mol = loadpdb 1PTR_POPC_ZAFF.pdb #Load the PDB file #this residue is split into PA and PC.という警告は無視
set mol box {66.2645315, 66.2645315, 100.353} #メモした値
bond mol.53.ZN mol.32.SG #Bond zinc ion with SG atom of residue CY2_32
bond mol.53.ZN mol.35.SG #Bond zinc ion with SG atom of residue CY2_35
bond mol.53.ZN mol.51.SG #Bond zinc ion with SG atom of residue CY2_51
bond mol.53.ZN mol.2.ND1 #Bond zinc ion with SG atom of residue HE1_2
bond mol.54.ZN mol.15.SG #Bond zinc ion with SG atom of residue CY2_15
bond mol.54.ZN mol.18.SG #Bond zinc ion with SG atom of residue CY2_18
bond mol.54.ZN mol.43.SG #Bond zinc ion with SG atom of residue CY2_43
bond mol.54.ZN mol.40.ND1 #Bond zinc ion with SG atom of residue HE1_40
savepdb mol 1PTR_POPC_ZAFF_leap.pdb #Save the pdb file
saveamberparm mol 1PTR_POPC_ZAFF_leap.prmtop 1PTR_POPC_ZAFF_leap.inpcrd #Save the topology and coordiante files
quit #Quit tleap
$ tleap -s -f tleap.in > tleap.outtleap.outを開いて,問題なく実行されていることを確認します.
Amber形式からGROMACS形式への変換とトポロジーファイルの修正
AMBER形式のファイルをAmberTools16付属のParmEdを使用してGROMACS形式に変換します.
以下のpythonスクリプトを実行してください.
ambertogromacs.py
import parmed as pmd
amber = pmd.load_file('1PTR_POPC_ZAFF_leap.prmtop',xyz='1PTR_POPC_ZAFF_leap.inpcrd')
amber.save('1PTR_POPC.top')
amber.save('1PTR_POPC.gro')
$ python ./ambertogromacs.py出力された1PTR_POPC.topを開き,リガンドの13位酸素原子に関するangleのパラメータを修正します (別記事を参照).
修正前:
28 52 53 1 59.09003 975.641856
27 52 53 1 59.09003 975.641856
48 52 53 1 115.68005 693.590048
修正後:
28 52 53 1 111.0 693.590048
27 52 53 1 118.7 693.590048
48 52 53 1 112.3 693.590048
また,PRB残基の電荷の総和が-0.004で0からずれているので,全ての原子に割り振って総和を0にします.
修正前 (先頭部分):
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
; residue 1 PRB rtp PRB q -0.0
1 o 1 PRB O3 1 -0.520100 16.0000 ; qtot -0.5201
2 c 1 PRB C3 2 0.549300 12.0100 ; qtot 0.0292
3 ce 1 PRB C2 3 -0.221400 12.0100 ; qtot -0.1922
4 c3 1 PRB C19 4 -0.038900 12.0100 ; qtot -0.2311
修正後:
[ atoms ]
; nr type resnr residue atom cgnr charge mass typeB chargeB massB
; residue 1 PRB rtp PRB q -0.0
1 o 1 PRB O3 1 -0.520032 16.0000 ; qtot -0.5201
2 c 1 PRB C3 2 0.549368 12.0100 ; qtot 0.0292
3 ce 1 PRB C2 3 -0.221332 12.0100 ; qtot -0.1922
4 c3 1 PRB C19 4 -0.038832 12.0100 ; qtot -0.2311
 Pymolで1PTR_DOPC.groを開いて,構造を確認します (右図).赤色の球はリン脂質のカルボニル酸素原子です.
Pymolで1PTR_DOPC.groを開いて,構造を確認します (右図).赤色の球はリン脂質のカルボニル酸素原子です.
GROMACSによるMDシミュレーション
Indexファイルとrestraintファイルの作成
charmm-gui.tgzを解凍したディレクトリ中のcharmm-gui/gromacsを見てみます.GROMACS用のmdpファイルとrestraintsファイルが入っています.CHARMM-GUI推奨の系の平衡化手順は以下の通りです.
! ! Setup Restraints for Protein and Lipids (see @liptype_restraint.str) ! ! Suggested Equilibration Scheme [Reducing Force Constants] ! (5 Cycles, 1 cycle = 50 - 100 ps ) !------------------------------------------------------------------------ ! 1 cycle 2 cycle 3 cycle 4 cycle 5 cycle 6 cycle !------------------------------------------------------------------------ ! BB 10.0 5.0 2.5 1.0 0.5 0.1 ! SC 5.0 2.5 1.0 0.5 0.1 0.0 ! tforce 2.5 1.0 0.5 0.0 0.0 0.0 ! mforce 2.5 0.0 0.0 0.0 0.0 0.0 ! ion 10.0 0.0 0.0 0.0 0.0 0.0 !------------------------------------------------------------------------ !
タンパク質の主鎖の重原子 (亜鉛を含む) およびリガンドの重原子の位置,側鎖の重原子の位置,リン脂質のリン原子のZ方向の位置,リン脂質のsn-1炭素–sn-3炭素–sn-2炭素–sn-2酸素の2面角,オレオイル基のcis二重結合の2面角に制限をかけて,ステップ毎に少しずつ制限を緩和していきます.CHARMM-GUIが出力したファイルとtleapで処理したファイルでは原子の順番が変わっているため,indexファイルとrestraintファイルを自分で作成します.
GROMACSにCY2残基とHE1残基をタンパク質と認識してもらうため,$GMXPREFIX/share/gromacs/top/residuetypes.datにこれらの残基を追加します.
CY2 Protein HE1 Protein ABU Protein
$ gmx make_ndx -f 1PTR_POPC.gro -o index.ndx
> 4 | 13 (主鎖とZNをmerge)
> 8 & !9 (側鎖の重原子を選択)
> 1 | 13 (タンパク質と亜鉛をmerge)
> name 34 RECEPTOR
> 34 | 14 (タンパク質-亜鉛とリガンドをmerge)
> name 35 PROT
> 15 | 16 | 17 (リン脂質同士をmerge)
> name 36 MEMB
> 26 | 27 | 28 (イオンと水をmerge)
> name 37 SOL_ION
> q主鎖+亜鉛用restraintファイルの作成:
$ gmx genrestr -f 1PTR_POPC.gro -n index.ndx -o PROT_rest.itp
Select a group: 34PROT_rest.itpを編集します.
修正前:
; position restraints for Backbone of GROningen MAchine for Chemical Simulation [ position_restraints ] ; i funct fcx fcy fcz 1 1 1000 1000 1000
修正後:
#ifdef STEP6_0 #ifdef STEP6_0 #define fc_bb 4000.0 #define fc_sc 2000.0 #endif #ifdef STEP6_1 #define fc_bb 4000.0 #define fc_sc 2000.0 #endif #ifdef STEP6_2 #define fc_bb 2000.0 #define fc_sc 1000.0 #endif #ifdef STEP6_3 #define fc_bb 1000.0 #define fc_sc 500.0 #endif #ifdef STEP6_4 #define fc_bb 500.0 #define fc_sc 200.0 #endif #ifdef STEP6_5 #define fc_bb 200.0 #define fc_sc 50.0 #endif #ifdef STEP6_6 #define fc_bb 50.0 #define fc_sc 0.0 #endif #ifdef STEP7 #define fc_bb 0.0 #define fc_sc 0.0 #endif [ position_restraints ] ; i funct fcx fcy fcz 1 1 fc_bb fc_bb fc_bb
側鎖用restraintファイルの作成:
$ gmx genrestr -f 1PTR_POPC.gro -n index.ndx -o SC_rest.itp
Select a group: 33SC_rest.itp中のforce constantsを1000からfc_scに全て置換し,[position_restraints] より後側をPROT_rest.itpのおしりにコピー&ペーストします.
さらに修正後のPROT_rest.itpの途中から:
801 1 fc_bb fc_bb fc_bb 802 1 fc_sc fc_sc fc_sc 803 1 fc_sc fc_sc fc_sc 6 1 fc_sc fc_sc fc_sc 7 1 fc_sc fc_sc fc_sc 13 1 fc_sc fc_sc fc_sc
リガンド用restraintファイルLIG_rest.itp:
#ifdef STEP6_0 #define fc_bb 4000.0 #endif #ifdef STEP6_1 #define fc_bb 4000.0 #endif #ifdef STEP6_2 #define fc_bb 2000.0 #endif #ifdef STEP6_3 #define fc_bb 1000.0 #endif #ifdef STEP6_4 #define fc_bb 500.0 #endif #ifdef STEP6_5 #define fc_bb 200.0 #endif #ifdef STEP6_6 #define fc_bb 50.0 #endif #ifdef STEP7 #define fc_bb 0.0 #endif [ position_restraints ] ; i funct fcx fcy fcz 1 1 fc_bb fc_bb fc_bb 2 1 fc_bb fc_bb fc_bb 3 1 fc_bb fc_bb fc_bb 4 1 fc_bb fc_bb fc_bb 8 1 fc_bb fc_bb fc_bb 10 1 fc_bb fc_bb fc_bb 12 1 fc_bb fc_bb fc_bb 13 1 fc_bb fc_bb fc_bb 15 1 fc_bb fc_bb fc_bb 18 1 fc_bb fc_bb fc_bb 19 1 fc_bb fc_bb fc_bb 20 1 fc_bb fc_bb fc_bb 24 1 fc_bb fc_bb fc_bb 26 1 fc_bb fc_bb fc_bb 27 1 fc_bb fc_bb fc_bb 28 1 fc_bb fc_bb fc_bb 29 1 fc_bb fc_bb fc_bb 33 1 fc_bb fc_bb fc_bb 39 1 fc_bb fc_bb fc_bb 40 1 fc_bb fc_bb fc_bb 42 1 fc_bb fc_bb fc_bb 43 1 fc_bb fc_bb fc_bb 48 1 fc_bb fc_bb fc_bb 49 1 fc_bb fc_bb fc_bb 52 1 fc_bb fc_bb fc_bb 53 1 fc_bb fc_bb fc_bb 54 1 fc_bb fc_bb fc_bb 55 1 fc_bb fc_bb fc_bb 56 1 fc_bb fc_bb fc_bb
リン脂質用restraintファイルPOPC_rest.itp:
#ifdef STEP6_0 #define fc_lpos 1000.0 #define fc_ldih 1000.0 #endif #ifdef STEP6_1 #define fc_lpos 1000.0 #define fc_ldih 1000.0 #endif #ifdef STEP6_2 #define fc_lpos 1000.0 #define fc_ldih 400.0 #endif #ifdef STEP6_3 #define fc_lpos 400.0 #define fc_ldih 200.0 #endif #ifdef STEP6_4 #define fc_lpos 200.0 #define fc_ldih 200.0 #endif #ifdef STEP6_5 #define fc_lpos 40.0 #define fc_ldih 100.0 #endif #ifdef STEP6_6 #define fc_lpos 0.0 #define fc_ldih 0.0 #endif #ifdef STEP7 #define fc_lpos 0.0 #define fc_ldih 0.0 #endif [ position_restraints ] 59 1 0.0 0.0 fc_lpos [ dihedral_restraints ] 50 55 53 82 1 120.0 2.5 fc_ldih 103 106 108 110 1 0.0 0.0 fc_ldih
各restraintファイルを読み込ませるために,1PTR_POPC.top中のタンパク質,リガンド,脂質それぞれの記述の最後に以下の記述を追加します.
794 798 796 797 4 180.00008 4.60240 2
#ifdef REST_ON
#include "./PROT_rest.itp"
#endif
[ moleculetype ]
; Name nrexcl
PRB 3
(略)
10 3 8 9 4 180.00008 4.60240 2
#ifdef REST_ON
#include "./LIG_rest.itp"
#endif
[ moleculetype ]
; Name nrexcl
system2 3
(略)
108 103 106 107 4 180.00008 4.60240 2
#ifdef REST_ON
#include "./POPC_rest.itp"
#endif
[ moleculetype ]
; Name nrexcl
K+ 3
系の平衡化
Energy Minimization
$ mkdir step6_0-6
$ cd step6_0-6
$ gmx grompp -f ../charmm-gui/gromacs/step6.0_minimization.mdp -c ../1PTR_POPC.gro -p ../1PTR_POPC.top -n ../index.ndx -o step6_0.tpr
$ gmx mdrun -v -deffnm step6_0
Pymolでstep6_0.groと1PTR_POPC.groを開き,タンパク質,リガンド,リン原子の位置等が制限されていることを確認します.
平衡化step 1 (NVT, 25 ps, time step = 1 fs)
$ gmx grompp -f ../charmm-gui/gromacs/step6.1_equilibration.mdp -c step6_0.gro -p ../1PTR_POPC.top -n ../index.ndx -o step6_1.tpr
$ gmx mdrun -v -deffnm step6_1
この25 psの計算に10分強かかります.膜の平衡化にかかる時間を考えると,以降の計算はスパコンで実行した方がよいですね.疎水性の環境でタンパク質とリガンドがどのように結合しているかのイメージはこれで出来ると思います.
(了)